
Abstract
Background
Lowe syndrome is characterized by the presence of congenital cataracts, psychomotor retardation, and dysfunctional proximal renal tubules. This study presents a case of an atypical phenotype, investigates the genetic characteristics of eight children diagnosed with Lowe syndrome in southern China, and performs functional analysis of the novel variants.
Methods
Whole-exome sequencing was conducted on eight individuals diagnosed with Lowe syndrome from three medical institutions in southern China. Retrospective collection and analysis of clinical and genetic data were performed, and functional analysis was conducted on the five novel variants.
Results
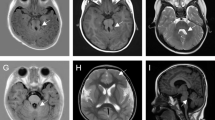
In our cohort, the clinical symptoms of the eight Lowe syndrome individuals varied. One patient was diagnosed with Lowe syndrome but did not present with congenital cataracts. Common features among all patients included cognitive impairment, short stature, and low molecular weight proteinuria. Eight variations in the OCRL gene were identified, encompassing three previously reported and five novel variations. Among the novel variations, three nonsense mutations were determined to be pathogenic, and two patients harboring novel missense variations of uncertain significance exhibited severe typical phenotypes. Furthermore, all novel variants were associated with altered protein expression levels and impacted primary cilia formation.
Conclusion
This study describes the first case of an atypical Lowe syndrome patient without congenital cataracts in China and performs a functional analysis of novel variants in the OCRL gene, thereby expanding the understanding of the clinical manifestations and genetic diversity associated with Lowe syndrome.
Graphical abstract

A higher resolution version of the Graphical abstract is available as Supplementary information




Similar content being viewed by others


Data availability
The corresponding author can provide the data supporting the findings of this study upon reasonable request.
References
Loi M (2006) Lowe syndrome. Orphanet J Rare Dis 1:16. https://doi.org/10.1186/1750-1172-1-16
Lowe CU, Terrey M, MacLachlan EA (1952) Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. AMA Am J Dis Child 83:164–184. https://doi.org/10.1001/archpedi.1952.02040060030004
Recker F, Zaniew M, Böckenhauer D, Miglietti N, Bökenkamp A, Moczulska A, Rogowska-Kalisz A, Laube G, Said-Conti V, Kasap-Demir B, Niemirska A, Litwin M, Siteń G, Chrzanowska KH, Krajewska-Walasek M, Sethi SK, Tasic V, Anglani F, Addis M, Wasilewska A, Szczepańska M, Pawlaczyk K, Sikora P, Ludwig M (2015) Characterization of 28 novel patients expands the mutational and phenotypic spectrum of Lowe syndrome. Pediatr Nephrol 30:931–943. https://doi.org/10.1007/s00467-014-3013-2
Charnas L, Bernar J, Pezeshkpour GH, Dalakas M, Harper GS, Gahl WA (1988) MRI findings and peripheral neuropathy in Lowe’s syndrome. Neuropediatrics 19:7–9. https://doi.org/10.1055/s-2008-1052393
Demmer LA, Wippold FJ 2nd, Dowton SB (1992) Periventricular white matter cystic lesions in Lowe (oculocerebrorenal) syndrome. A new MR finding. Pediatr Radiol 22:76–77. https://doi.org/10.1007/BF02011619
Bockenhauer D, Bokenkamp A, van’t Hoff W, Levtchenko E, Kist-van Holthe JE, Tasic V, Ludwig M (2008) Renal phenotype in Lowe syndrome: a selective proximal tubular dysfunction. Clin J Am Soc Nephrol 3:1430–1436. https://doi.org/10.2215/CJN.00520108
Cau M, Addis M, Congiu R, Meloni C, Cao A, Santaniello S, Loi M, Emma F, Zuffardi O, Ciccone R, Sole G, Melis MA (2006) A locus for familial skewed X chromosome inactivation maps to chromosome Xq25 in a family with a female manifesting Lowe syndrome. J Hum Genet 51:1030–1036. https://doi.org/10.1007/s10038-006-0049-6
Hodgson SV, Heckmatt JZ, Hughes E, Crolla JA, Dubowitz V, Bobrow M (1986) A balanced de novo X/autosome translocation in a girl with manifestations of Lowe syndrome. Am J Med Genet 23:837–847. https://doi.org/10.1002/ajmg.1320230311
Lin T, Lewis RA, Nussbaum RL (1999) Molecular confirmation of carriers for Lowe syndrome. Ophthalmology 106:119–122. https://doi.org/10.1016/S0161-6420(99)90012-X
Pirruccello M, De Camilli P (2012) Inositol 5-phosphatases: insights from the Lowe syndrome protein OCRL. Trends Biochem Sci 37:134–143. https://doi.org/10.1016/j.tibs.2012.01.002
Recker F, Reutter H, Ludwig M (2013) Lowe syndrome/Dent-2 disease: a comprehensive review of known and novel aspects. J Pediatr Genet 2:53–68. https://doi.org/10.3233/PGE-13049
Mehta ZB, Pietka G, Lowe M (2014) The cellular and physiological functions of the Lowe syndrome protein OCRL1. Traffic 15:471–487. https://doi.org/10.1111/tra.12160
Vicinanza M, Di Campli A, Polishchuk E, Santoro M, Di Tullio G, Godi A, Levtchenko E, De Leo MG, Polishchuk R, Sandoval L, Marzolo MP, De Matteis MA (2011) OCRL controls trafficking through early endosomes via PtdIns4,5P2-dependent regulation of endosomal actin. EMBO J 30:4970–4985. https://doi.org/10.1038/emboj.2011.354
Festa BP, Berquez M, Gassama A, Amrein I, Ismail HM, Samardzija M, Staiano L, Luciani A, Grimm C, Nussbaum RL, De Matteis MA, Dorchies OM, Scapozza L, Wolfer DP, Devuyst O (2019) OCRL deficiency impairs endolysosomal function in a humanized mouse model for Lowe syndrome and Dent disease. Hum Mol Genet 28:1931–1946. https://doi.org/10.1093/hmg/ddy449
Karabiyik C, Son SM, Rubinsztein DC (2021) Lysosome positioning and mTOR activity in Lowe syndrome. EMBO Rep 22:53232. https://doi.org/10.15252/embr.202153232
Hichri H, Rendu J, Monnier N, Coutton C, Dorseuil O, Poussou RV, Baujat G, Blanchard A, Nobili F, Ranchin B, Remesy M, Salomon R, Satre V, Lunardi J (2011) From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum Mutat 32:379–388. https://doi.org/10.1002/humu.21391
Pasternack SM, Böckenhauer D, Refke M, Tasic V, Draaken M, Conrad C, Born M, Betz RC, Reutter H, Ludwig M (2013) A premature termination mutation in a patient with Lowe syndrome without congenital cataracts: dropping the “O” in OCRL. Klin Padiatr 225:29–33. https://doi.org/10.1055/s-0032-1321900
Bökenkamp A, Ludwig M (2016) The oculocerebrorenal syndrome of Lowe: an update. Pediatr Nephrol 31:2201–2212. https://doi.org/10.1007/s00467-016-3343-3
Zhang Y, Deng L, Chen X, Hu Y, Chen Y, Chen K, Zhou J (2021) Novel pathogenic OCRL mutations and genotype-phenotype analysis of Chinese children affected by oculocerebrorenal syndrome: two cases and a literature review. BMC Med Genomics 14:219. https://doi.org/10.1186/s12920-021-01069-9
Hoopes RR Jr, Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J, Simckes A, Tasic V, Toenshoff B, Suchy SF, Nussbaum RL, Scheinman SJ (2005) Dent disease with mutations in OCRL1. Am J Hum Genet 76:260–267. https://doi.org/10.1086/427887
Utsch B, Bökenkamp A, Benz MR, Besbas N, Dötsch J, Franke I, Fründ S, Gok F, Hoppe B, Karle S, Kuwertz-Bröking E, Laube G, Neb M, Nuutinen M, Ozaltin F, Rascher W, Ring T, Tasic V, van Wijk JA, Ludwig M (2006) Novel OCRL1 mutations in patients with the phenotype of Dent disease. Am J Kidney Dis 48:942.e1-e14. https://doi.org/10.1053/j.ajkd.2006.08.018
Bökenkamp A, Böckenhauer D, Cheong HI, Hoppe B, Tasic V, Unwin R, Ludwig M (2009) Dent-2 disease: a mild variant of Lowe syndrome. J Pediatr 155:94–99. https://doi.org/10.1016/j.jpeds.2009.01.049
Böckenhauer D, Bökenkamp A, Nuutinen M, Unwin R, Van’t Hoff W, Sirimanna T, Vrljicak K, Ludwig M (2012) Novel OCRL mutations in patients with Dent-2 disease. J Pediatr Genet 1:15–23. https://doi.org/10.3233/PGE-2012-005
Li H, Ji CY, Zong XN, Zhang YQ (2009) Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years. Zhonghua er ke za zhi (Chin J Pediatr) 47:487–492
Li H, Ji CY, Zong XN, Zhang YQ (2009) Body mass index growth curves for Chinese children and adolescents aged 0 to 18 years. Zhonghua er ke za zhi (Chin J Pediatr) 47:493–498
(2018) A national survey on physical growth and development of children under seven years of age in nine cities of China in 2015. Zhonghua er ke za zhi (Chin J Pediatr) 56:192–199. https://doi.org/10.3760/cma.j.issn.0578-1310.2018.03.008
Matos V, van Melle G, Boulat O, Markert M, Bachmann C, Guignard JP (1997) Urinary phosphate/creatinine, calcium/creatinine, and magnesium/creatinine ratios in a healthy pediatric population. J Pediatr 131:252–257. https://doi.org/10.1016/s0022-3476(97)70162-8
Kruse K, Kracht U, Göpfert G (1982) Renal threshold phosphate concentration (TmPO4/GFR). Arch Dis Child 57:217–223. https://doi.org/10.1136/adc.57.3.217
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Coon BG, Hernandez V, Madhivanan K, Mukherjee D, Hanna CB, Barinaga-Rementeria Ramirez I, Lowe M, Beales PL, Aguilar RC (2012) The Lowe syndrome protein OCRL1 is involved in primary cilia assembly. Hum Mol Genet 21:1835–1847. https://doi.org/10.1093/hmg/ddr615
Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X (2009) DOG 1.0: illustrator of protein domain structures. Cell Res 19:271–273. https://doi.org/10.1038/cr.2009.6
Charnas LR, Bernardini I, Rader D, Hoeg JM, Gahl WA (1991) Clinical and laboratory findings in the oculocerebrorenal syndrome of Lowe, with special reference to growth and renal function. N Engl J Med 324:1318–1325. https://doi.org/10.1056/NEJM199105093241904
Gobernado JM, Lousa M, Gimeno A, Gonsalvez M (1984) Mitochondrial defects in Lowe’s oculocerebrorenal syndrome. Arch Neurol 41:208–209. https://doi.org/10.1001/archneur.1984.04050140106037
Park E, Choi HJ, Lee JM, Ahn YH, Kang HG, Choi YM, Park SJ, Cho HY, Park YH, Lee SJ, Ha IS, Cheong HI (2014) Muscle involvement in Dent disease 2. Pediatr Nephrol 29:2127–2132. https://doi.org/10.1007/s00467-014-2841-4
Lewis RA, Nussbaum RL, Brewer ED (2001) Lowe syndrome. In: Adam MP, Feldman J, Mirzaa GM et al (eds) GeneReviews®. University of Washington, Seattle
Kleta R (2008) Fanconi or not Fanconi? Lowe syndrome revisited. Clin J Am Soc Nephrol 3:1244–1245. https://doi.org/10.2215/CJN.02880608
Nakano E, Yoshida A, Miyama Y, Yabuuchi T, Kajiho Y, Kanda S, Miura K, Oka A, Harita Y (2020) Incomplete cryptic splicing by an intronic mutation of OCRL in patients with partial phenotypes of Lowe syndrome. J Hum Genet 65:831–839. https://doi.org/10.1038/s10038-020-0773-3
Tosetto E, Addis M, Caridi G, Meloni C, Emma F, Vergine G, Stringini G, Papalia T, Barbano G, Ghiggeri GM, Ruggeri L, Miglietti N, D’Angelo A, Melis MA, Anglani F (2009) Locus heterogeneity of Dent’s disease: OCRL1 and TMEM27 genes in patients with no CLCN5 mutations. Pediatr Nephrol 24:1967–1973. https://doi.org/10.1007/s00467-009-1228-4
Ma Z, Zhu P, Shi H, Guo L, Zhang Q, Chen Y, Chen S, Zhang Z, Peng J, Chen J (2019) PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 568:259–263. https://doi.org/10.1038/s41586-019-1057-y
El-Brolosy MA, Kontarakis Z, Rossi A, Kuenne C, Günther S, Fukuda N, Kikhi K, Boezio G, Takacs CM, Lai SL, Fukuda R, Gerri C, Giraldez AJ, Stainier D (2019) Genetic compensation triggered by mutant mRNA degradation. Nature 568:193–197. https://doi.org/10.1038/s41586-019-1064-z
Bothwell SP, Farber LW, Hoagland A, Nussbaum RL (2010) Species-specific difference in expression and splice-site choice in Inpp5b, an inositol polyphosphate 5-phosphatase paralogous to the enzyme deficient in Lowe syndrome. Mamm Genome 21:458–466. https://doi.org/10.1007/s00335-010-9281-7
Cooper DN, Ball EV, Stenson PD, Phillips AD, Evans K, Heywood S, Hayden MJ, Azevedo L, Mort ME, Hussain (2020) The human gene mutation database. http://www.hgmd.cf.ac.uk/ac/index.php. Accessed 30 April 2023
Lin T, Orrison BM, Leahey AM, Suchy SF, Bernard DJ, Lewis RA, Nussbaum RL (1997) Spectrum of mutations in the OCRL1 gene in the Lowe oculocerebrorenal syndrome. Am J Hum Genet 60:1384–1388. https://doi.org/10.1086/515471
Satre V, Monnier N, Berthoin F, Ayuso C, Joannard A, Jouk PS, Lopez-Pajares I, Megabarne A, Philippe HJ, Plauchu H, Torres ML, Lunardi J (1999) Characterization of a germline mosaicism in families with Lowe syndrome, and identification of seven novel mutations in the OCRL1 gene. Am J Hum Genet 65:68–76. https://doi.org/10.1086/302443
Monnier N, Satre V, Lerouge E, Berthoin F, Lunardi J (2000) OCRL1 mutation analysis in French Lowe syndrome patients: implications for molecular diagnosis strategy and genetic counseling. Hum Mutat 16:157–165. https://doi.org/10.1002/1098-1004(200008)16:2%3c157::AID-HUMU8%3e3.0.CO;2-9
Addis M, Loi M, Lepiani C, Cau M, Melis MA (2004) OCRL mutation analysis in Italian patients with Lowe syndrome. Hum Mutat 23:524–525. https://doi.org/10.1002/humu.9239
Cho HY, Lee BH, Choi HJ, Ha IS, Choi Y, Cheong HI (2008) Renal manifestations of Dent disease and Lowe syndrome. Pediatr Nephrol 23:243–249. https://doi.org/10.1007/s00467-007-0686-9
Kim HK, Kim JH, Kim YM, Kim GH, Lee BH, Choi JH, Yoo HW (2014) Lowe syndrome: a single center’s experience in Korea. Korean J Pediatr 57:140–148. https://doi.org/10.3345/kjp.2014.57.3.140
Lowe M (2005) Structure and function of the Lowe syndrome protein OCRL1. Traffic 6:711–719. https://doi.org/10.1111/j.1600-0854.2005.00311.x
Choudhury R, Diao A, Zhang F, Eisenberg E, Saint-Pol A, Williams C, Konstantakopoulos A, Lucocq J, Johannes L, Rabouille C, Greene LE, Lowe M (2005) Lowe syndrome protein OCRL1 interacts with clathrin and regulates protein trafficking between endosomes and the trans-Golgi network. Mol Biol Cell 16:3467–3479. https://doi.org/10.1091/mbc.e05-02-0120
Bacic D, Lehir M, Biber J, Kaissling B, Murer H, Wagner CA (2006) The renal Na+/phosphate cotransporter NaPi-IIa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone. Kidney Int 69:495–503. https://doi.org/10.1038/sj.ki.5000148
Erdmann KS, Mao Y, McCrea HJ, Zoncu R, Lee S, Paradise S, Modregger J, Biemesderfer D, Toomre D, De Camilli P (2007) A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev Cell 13:377–390. https://doi.org/10.1016/j.devcel.2007.08.004
Lichter-Konecki U, Farber LW, Cronin JS, Suchy SF, Nussbaum RL (2006) The effect of missense mutations in the RhoGAP-homology domain on ocrl1 function. Mol Genet Metab 89:121–128. https://doi.org/10.1016/j.ymgme.2006.04.005
Prosseda PP, Luo N, Wang B, Alvarado JA, Hu Y, Sun Y (2017) Loss of OCRL increases ciliary PI(4,5)P(2) in Lowe oculocerebrorenal syndrome. J Cell Sci 130:3447–3454. https://doi.org/10.1242/jcs.200857
Dambournet D, Machicoane M, Chesneau L, Sachse M, Rocancourt M, El Marjou A, Formstecher E, Salomon R, Goud B, Echard A (2011) Rab35 GTPase and OCRL phosphatase remodel lipids and F-actin for successful cytokinesis. Nat Cell Biol 13:981–988. https://doi.org/10.1038/ncb2279
Ramadesikan S, Skiba L, Lee J, Madhivanan K, Sarkar D, De La Fuente A, Hanna CB, Terashi G, Hazbun T, Kihara D, Aguilar RC (2021) Genotype & phenotype in Lowe syndrome: specific OCRL1 patient mutations differentially impact cellular phenotypes. Hum Mol Genet 30:198–212. https://doi.org/10.1093/hmg/ddab025
Wu G, Zhang W, Na T, Jing H, Wu H, Peng JB (2012) Suppression of intestinal calcium entry channel TRPV6 by OCRL, a lipid phosphatase associated with Lowe syndrome and Dent disease. Am J Physiol Cell Physiol 302:C1479-1491. https://doi.org/10.1152/ajpcell.00277.2011
Luo N, Conwell MD, Chen X, Kettenhofen CI, Westlake CJ, Cantor LB, Wells CD, Weinreb RN, Corson TW, Spandau DF, Joos KM, Iomini C, Obukhov AG, Sun Y (2014) Primary cilia signaling mediates intraocular pressure sensation. Proc Natl Acad Sci U S A 111:12871–12876. https://doi.org/10.1073/pnas.1323292111
Suchy SF, Cronin JC, Nussbaum RL (2009) Abnormal bradykinin signalling in fibroblasts deficient in the PIP(2) 5-phosphatase, ocrl1. J Inherit Metab Dis 32:280–288. https://doi.org/10.1007/s10545-009-1058-3
Gupta PD, Johar K, Vasavada A (2004) Causative and preventive action of calcium in cataracto-genesis. Acta Pharmacol Sin 25:1250–1256
Montjean R, Aoidi R, Desbois P, Rucci J, Trichet M, Salomon R, Rendu J, Fauré J, Lunardi J, Gacon G, Billuart P, Dorseuil O (2015) OCRL-mutated fibroblasts from patients with Dent-2 disease exhibit INPP5B-independent phenotypic variability relatively to Lowe syndrome cells. Hum Mol Genet 24:994–1006. https://doi.org/10.1093/hmg/ddu514
Luo N, West CC, Murga-Zamalloa CA, Sun L, Anderson RM, Wells CD, Weinreb RN, Travers JB, Khanna H, Sun Y (2012) OCRL localizes to the primary cilium: a new role for cilia in Lowe syndrome. Hum Mol Genet 21:3333–3344. https://doi.org/10.1093/hmg/dds163
Rbaibi Y, Cui S, Mo D, Carattino M, Rohatgi R, Satlin LM, Szalinski CM, Swanhart LM, Fölsch H, Hukriede NA, Weisz OA (2012) OCRL1 modulates cilia length in renal epithelial cells. Traffic 13:1295–1305. https://doi.org/10.1111/j.1600-0854.2012.01387.x
Luo N, Kumar A, Conwell M, Weinreb RN, Anderson R, Sun Y (2013) Compensatory role of inositol 5-phosphatase INPP5B to OCRL in primary cilia formation in oculocerebrorenal syndrome of Lowe. PLoS One 8:e66727. https://doi.org/10.1371/journal.pone.0066727
Acknowledgements
The authors express their gratitude to the participating families for their involvement in this study. Appreciation is also extended to the medical detection departments of hospitals for their valuable support in conducting imaging and biochemical examinations.
Funding
This work was supported by the Enterprise Joint Fund of Guangdong Basic and Applied Basic Research Fund (Public Health and Medical Health) (grant number 2021A1515220168).
Author information
Authors and Affiliations
Contributions
CZ, LL, and WZ designed the study. RD, TL, AX, YH, HM, DT, XH, RZ, CL, WZ, LL, and CZ obtained clinical information. RD, CZH, SC, YL, YS, and YC conducted the experiments. RD and CZ performed data analysis. RD authored the paper. RD and CZ reviewed and revised the paper. All authors approved of the manuscript prior to submission.
Corresponding author
Ethics declarations
Ethics approval
The research involving human participants underwent ethical review and received approval from the Institutional Review Board of Guangzhou Women and Children’s Medical Center. Written informed consent was obtained from the legal guardian of each participant prior to their involvement in the study.
Consent to participate
The participants’ guardians provided written consent for their participation in the study.
Consent for publication
Each author on this list contributed to the writing or revising of the final manuscript and gave their ultimate approval for publication.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rong Du, Chengcheng Zhou, and Shehong Chen contributed to the work equally and should be regarded as co-first authors.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Du, R., Zhou, C., Chen, S. et al. Atypical phenotypes and novel OCRL variations in southern Chinese patients with Lowe syndrome. Pediatr Nephrol (2024). https://doi.org/10.1007/s00467-024-06356-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00467-024-06356-y