
Abstract
Immiscibility between water and hydrogen-rich fluids may be responsible for the formation of super-reduced mineral assemblages and for the early oxidation of Earth´s upper mantle. In the current study, we present new data on the critical curve in the H2-H2O system to 1400 ℃ and 4 GPa. We utilized a synthetic fluid inclusion method to trap fluids at high P–T conditions within quartz and olivine crystals. Experiments were performed in a piston-cylinder type apparatus, employing a double-capsule technique. The inner capsule contained the crystal and fluids of interest, while the outer served as oxygen fugacity buffer, maintaining f(O2) at the iron-wüstite (Fe-FeO) equilibrium. Our results suggest that below ~ 2.5 GPa, the critical curve has a mostly linear slope of 200 ℃/GPa, while at more elevated pressure it becomes significantly steeper (400 ℃/GPa). This implies that in most of the modern, reduced upper mantle, water and hydrogen are immiscible, while localized heating events, such as rising plumes, may close the miscibility gap. The steep increase of the critical curve at high pressure observed in this study implies that even for very hot geotherms in the early Archean or the late Hadean, H2-H2O immiscibility likely occurred in the deeper parts of the upper mantle, thus making a plausible case for rapid H2 loss as a mechanism of early mantle oxidation. To constrain the geochemical fingerprint of this process, we performed a series of element partitioning experiments to unravel how the H2-H2O unmixing may affect element transfer. Noble gases such as Xe as well as methane are preferentially incorporated in the hydrogen-rich phase, with a XeH2O/XeH2 ratio of ~ 8. This partitioning pattern may, for example, explain the underabundance of Xe isotopes produced by fission of Pu in the mantle. These Xe isotopes may have been removed by a primordial H2-H2O unmixing event during the early stages of planetary evolution.
Similar content being viewed by others

Introduction
Hydrogen speciation in the interior of planets depends on oxidation state. For example, in Earth’s uppermost lithospheric mantle, oxygen fugacity f(O2) ranges from + 2 to -3 log units relative to the quartz-fayalite-magnetite buffer (ΔQFM) (Ballhaus 1993; Frost and McCammon 2008). Such conditions are typically considered as relatively oxidizing for the mantle, with the primary hydrogen species being molecular H2O or bound hydroxyl groups (OH−). At greater depths and pressures, exceeding 200 km and 5–7 GPa, respectively, oxygen fugacity evolves towards more reducing values, approaching the iron-wüstite (IW) buffer with relatively little further deviation (1–1.5 log units) (Frost and McCammon 2008). As the environment becomes more reducing, the fraction of water in fluids decreases and molecular hydrogen H2 as well as methane CH4 become the primary H-species. Recent studies indicate that in the more reduced parts of the mantle, H2 may not only be present in a free fluid phase, but also as dissolved species in minerals and melts (Hirschmann et al. 2012; Yang et al. 2016).
While the depth-dependent fugacity profile described above holds true for the majority of the contemporary mantle environments, there may be local, though geologically significant exceptions. Enigmatic super-reducing domains contain mineral assemblages recording log f(O2) far below the IW buffer. A characteristic phase, most commonly cited as an indicator of such super-reducing environments, is moissanite (SiC). The formation of SiC requires extremely low oxygen fugacities with log f(O2) approximately 5–8 units below the IW buffer (e.g. Golubkova et al. 2016; Ulmer et al. 1998). In the past, it was sometimes questioned, whether the moissanite in mantle samples is actually a natural phase or a synthetic contamination. However, SiC has now been discovered in situ in a wide range of geological settings, including kimberlites and lamproites (Kaminsky 2012; Shiryaev et al. 2011), ophiolites (Golubkova et al. 2016; Pujol-Solà et al. 2018; Trumbull et al. 2009), alkaline basalt tuffs (Dobrzhinetskaya et al. 2018), and syenites (Nazzareni et al. 2019). In such super-reducing mineral assemblages, moissanite is commonly associated with native iron, silicon, various alloys, wüstite, phosphides and nitrides. Traditionally, these phases were thought to originate at great depth (< 200–250 km), where conditions may perhaps be locally more reducing. While this may be so in some cases, Bali et al. (2013) suggested that super-reducing conditions can emerge even at 100–200 km depth from the percolation of hydrogen-rich fluids, produced via unmixing in the H2-H2O system.
At present, super-reducing domains are uncommon, since Earth’s upper mantle is quite oxidized and comparatively cold (e.g. Abbott et al. 1994; Herzberg et al. 2010). However, at the dawn of the Archean and before, its oxidation and thermal state may have been dramatically different. This is especially true for the first two hundred million years of Earth’s history. During and immediately after core formation, as indicated by existing models (Rubie et al. 2011), Earth’s interior likely was very reduced, with f(O2) being several log units below the IW buffer. Throughout this stage, our planet, as well as Mars and Venus, may have had a hydrogen-rich atmosphere, supported by magma ocean outgassing (Lammer et al. 2018). However, for Earth, the upper mantle remained reduced at most for about 200 Myr. Based on the oxygen fugacity estimates of Trail et al. (2011), the redox state of the upper mantle was already close to that prevailing today at 4350 Myr before present. An early oxidation of the upper mantle has been noted for Mars as well, suggesting that it may be a common phase of the evolution of the terrestrial planets (Tuff et al. 2013).
The short timescale of the oxidation event points towards a fast process responsible for it. While the exact mechanism is still under discussion (Armstrong et al. 2019; Williams et al. 2012), the emergence of the miscibility gap between water and hydrogen upon cooling of the mantle was considered as a plausible cause (Bali et al. 2013). Global degassing of a highly mobile and buoyant H2-rich phase could have rapidly oxidized the remaining mantle. Moreover, it would certainly have affected the mantle abundances of other elements, especially noble gases, such as Xe or Ar. An early loss of radiogenic 129Xe and of Xe isotopes produced by Pu fission has indeed been reported (Kunz et al. 1998), but linking it to H2-H2O unmixing requires the knowledge of partition coefficients between water and hydrogen at reduced upper mantle conditions that are currently unknown.
In this paper, we present new data on the position of the critical curve in the H2-H2O system from 1 to 4 GPa, which approximately doubles the data coverage from the previous study of Bali et al. (2013). Moreover, we report the first experiments on element partitioning between the two coexisting fluid phases. The data will be applied to better understand the formation of super-reducing domains, the processes of mantle degassing during the first stages of Earth´s history, and the causes for the rapid loss of Xe from the early Earth.
Experimental methods
Starting material and capsule preparation
For trapping fluid inclusions, small cylinders (diameter 2.2 mm, length 1 mm) were cut out of bulk pieces of clear, natural quartz (Brazil), natural olivine (Dac Lac, Vietnam), and synthetic forsterite. Cylinders were thermally cracked by heating them to 1000 °C in a furnace and then immediately dropping them into distilled water. For experiments at pressures below 2.5 GPa, quartz was used to trap the inclusions, while forsterite was chosen for higher pressures. This change of the host crystal is necessary because of the quartz–coesite transition, which transforms a single crystal of quartz into a polycrystalline aggregate of coesite unsuitable for trapping inclusions (Lathe et al. 2005).
A double-capsule technique (for detailed discussion, see Chou 1987) was used to buffer the oxygen fugacity at the Fe-FeO equilibrium, to contain hydrogen during the experiment, and to protect the crystals from Fe contamination. In several early test runs for measuring the critical curve, the forsterite crystal core was loaded directly into the buffering mixture (Fe + FeO), without the inner capsule. We found out that with olivine, this method cannot be utilized at temperatures above 1100–1200 °C in the selected pressure range due to the precipitation of Fe-rich brucite and pure iron that complicate the analysis of H2 and H2O in the fluid inclusions. Two low-temperature (900 and 1000 °C) 3 GPa samples produced in this way were used in the present study, as they show no signs of the aforementioned additional solid phases. For the rest of the experiments in this study, a double-capsule was used, regardless of the crystal type. Depending on the purpose of the run–locating the critical curve or element partitioning experiments–only the contents of the inner capsule differed, with the rest of the assembly remaining unchanged and being generally similar to the technique utilized by Bali et al. (2013).
For experiments to determine the position of the critical curve, the inner Pt95Rh5 capsule (outer diameter 2.5 mm, inner diameter 2.3 mm, length 5–6 mm) contained only the crystal cylinder, with the rest being filled up with ultrapure water (Millipore, σ = 0.06 μS) (Fig. 1a). For element partitioning experiments the capsule was filled from bottom to top in the following sequence: A layer of Ca(OH)2 (Merck, > 96%), XeF2 crystals (Alfa Aesar, 99.5%), a layer of synthetic diopside-glass powder doped with about 25 trace elements at the 0.5–1 wt. % level (sample MORB2-2D2, Table S8 in supplementary file 1, Rustioni et al. 2019), Au foil (24 carat, Noris Blattgoldfabrik), the crystal cylinder, and ~ 10 μl of water containing 1000 ppm of both Rb and Cs (Fig. 1b). The solution, replacing pure water in this type of capsule, was prepared by the addition of RbCl (chemPUR, 99.9%) and CsCl (chemPUR, 99.99%) to the distilled water. Here, Xe and Rb/Cs were introduced for use as internal standards during later analysis, assuming that they would be incorporated primarily into the H2 and H2O phases, respectively. While adding Xe in the form of the solid XeF2 is convenient, the release of fluorine during the decomposition of XeF2 into Xe and F2 upon heating is undesired. The Ca(OH)2 layer binds this excess fluorine in the form of CaF2. This prevents the formation of HF as an unwanted component in the fluid. The thin gold membrane separated H2O from XeF2 during capsule preparation, in order to avoid premature decomposition by hydrolysis (XeF2 + H2O → Xe + 0.5O2 + 2HF). After the addition of the crystal and water or solution the capsule was arc-welded. Following the welding procedure, the inner capsule was inserted into the outer capsule machined of Fe (GoodFellow, 99.8%, outer diameter 5 mm, inner diameter 3 mm, length 10 mm), together with a mixture of distilled water and FeO (Sigma-Aldrich, 99.7%) that ensured Fe-FeO buffer conditions (Fig. 1c). This outer capsule was closed by inserting the lid and was mechanically sealed on compression.

Design of the double-capsules used for experiments. a and b show details of inner capsules, c shows the entire double-capsule. For experiments used to locate the critical curve, inner capsules of type a were used, while type b was used for element partitioning experiments. Diagrams are not to scale
Piston-cylinder experiments
Experiments were carried out in an end-loaded piston-cylinder apparatus using ½’’ talc/pyrex assemblies with a tapered graphite heater. A friction correction of 18% was applied to the nominal pressure, according to a calibration by the quartz–coesite transition at 790 °C (Bose and Ganguly 1995). Temperature was measured with S-type thermocouples (Pt–Pt90Rh10) below 1300 °C, and with D-type thermocouples (W97Re3–W75Re25) at higher temperatures. To ensure minimal secondary alteration and recrystallization of the inclusions, run durations varied between 24 and 5 h (for exact run durations see Table 1). At the end of the run, 25–30% of the pressure was released over the period of 10–30 min, followed by several hours of continuous decompression to ambient conditions. Simultaneously with the initial pressure release, the runs were quenched at ~ 100 °C/minute. According to previous research performed at similar conditions, this approach allows to preserve inclusions and minimizes secondary changes (Bali et al. 2013; Tingle et al. 1992). For pressures ≥ 3 GPa, the decompression P–T paths were slightly adjusted to avoid ice formation, with an addition of a low temperature heating step (~ 50–75 °C, max time–2 h) during the final pressure release.
Sample preparation and Raman investigation
Capsules were cut open after the runs, crystals retrieved, mounted in epoxy resin and doubly polished to a thickness of 100–200 μm. Run products were initially investigated and inclusions were mapped out with a Leica DMLP polarization microscope with objectives ranging from 5 × to 50x. Photographs were taken with a Canon EOS D60 camera, mounted on the microscope. Raman spectra of coexisting fluid phases were collected with a Horiba Labram 800 HR UV confocal Raman spectrometer using the 514 nm line of an argon laser at 200 mW output power, a 1800 mm−1 grating, a two-dimensional CCD detector, and a confocal pinhole of 100 μm with a 50 × long-distance objective.
LA-ICP-MS analyses
Prior to the LA-ICP-MS measurements, fluid inclusions were selected under the microscope, photographed, and the presence of H2 and H2O-rich phases were confirmed with Raman spectroscopy. LA-ICP-MS analyses were performed with a 193 nm ArF Excimer laser ablation unit (GeolasPro system, Coherent, USA) connected to an Elan DRC-e quadrupole ICP-MS (Perkin Elmer, Canada). The sample chamber was flushed with He at 0.4 l/min, to which 5 ml/min of H2 was added on the way to the ICP-MS machine. Inclusions and standards were ablated with a repetition rate of 10 Hz, with the energy for standards being 90 mJ and for the inclusions 180 mJ. Laser diameter varied based on the size of the inclusion from 8 to 25 μm. NIST SRM 610 glass was used as external standard for all analyzed elements except for Xe, for which a separate glass standard was prepared as detailed below. Analyzed isotopes were 131Xe, 132Xe, 85Rb, 7Li, 11B, 27Al, 90Zr, 133Cs.
As there are no commercially available EPMA or ICP-MS standards for Xe, a new reference material was synthesized. A Xe-bearing glass was produced from a mixture of XeF2 and powdered natural Armenian obsidian (OA-1; Wu et al. 2022) as starting materials. Xe was added as XeF2 to yield a concentration of 1 wt.% Xe (Schmidt and Keppler 2002; Shibata et al. 1998). Those compounds were mixed, loaded into a Pt capsule, and heated for 1 h at 1500 °C and 2 GPa in a piston-cylinder apparatus (½’’ talc/pyrex assembly) and quenched. Optical examination showed no inhomogeneity.
Although Xe, Rb and Cs were originally added to the fluid to serve as internal standard to quantify the signals obtained from H2-rich and H2O-rich inclusions, respectively, quantification of the H2-rich inclusions turned out to be impractical. The results will thus be discussed solely based on relative and absolute signal intensities without any use of internal standards.
Results
Position of the critical curve in the H2-H2O system
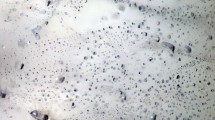
Fluid inclusion-capturing experiments were performed for 30 individual pressure–temperature conditions to investigate the location of critical curve in the H2-H2O system from 1 to 4 GPa (Fig. 2, Table 1). Three types of inclusions can be observed in the samples: Darker, nearly pure H2 inclusions with strong optical contrast to the host crystal, and lighter H2O-rich inclusions with weak optical contrast, as well as inclusions containing both phases at the same time.

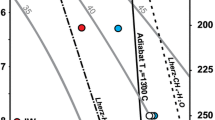
Location of the critical curve in the H2-H2O system (green line) according to our experimental data (green symbols). Open squares–only mixed water-hydrogen inclusions are present (full miscibility), filled circles indicate the presence of pure water and pure hydrogen inclusions (immiscibility). The critical curve from Bali et al. (2013) is plotted as orange line with their experimental data being symbols of the same color. The lavender line is a critical curve established by Seward and Franck (1981) and the blue line is a critical curve from a recent computational (Gibbs-ensemble Monte Carlo simulations) study by Bergermann et al. (2021). The dashed, dark grey line separates experiments performed with quartz versus ones performed with olivine, both in this work and in (Bali et al. 2013)
Depending on the phase assemblages present in the inclusions, two distinctively different groups of experimental run products can be identified. The first group, produced at lower temperatures, contains all three types of inclusions–with nearly pure H2, H2O, and their mixture. Following Bali et al. (2013), we interpret this as entrapment within the region of H2-H2O immiscibility (Fig. 3a). The second group, synthesized at higher temperatures, contains only mixed H2-H2O inclusions which we interpret as evidence for full miscibility between those two components at entrapment conditions (Fig. 3b).

Synthetic fluid inclusions formed a in quartz at 1.5 GPa and 700 °C, with Raman spectra of the vapor bubble in a water-rich inclusion shown in orange, liquid water in blue, and hydrogen gas in green, and b in synthetic forsterite at 2.5 GPa and 1050 °C, with Raman spectra of water shown in orange, and of hydrogen in green. The spectra in Figure b were baseline-corrected, the image of inclusions in the same figure is a combination of two photographs made at different focal depths
Pure liquid water inclusions without any additional phases are observed only in samples synthesized at pressures above ~ 2.5 GPa. Below this value (e.g. supplementary file 2, Figure S1a,c,e), the change in fluid density during cooling causes nucleation of a water vapour bubble. This vapour bubble commonly contains trace amounts of hydrogen since the solubility of hydrogen in water increases as the critical curve is approached from within the immiscibility region. At pressures < 2 GPa, the temperature range with notable H2 solubility appears to be larger (~ 100 ˚C or even more) than at higher pressures, as indicated by detectable amounts of hydrogen in water inclusions (Fig. 3a). The fluid inclusions in Fig. 3a were synthesized in quartz at 1.5 GPa and 800 °C, i.e., at conditions close to critical curve, but still in the immiscibility region. Two phases can be seen in the water-rich inclusion: liquid water with minor amounts of dissolved hydrogen (blue spectrum), and water vapour (orange spectrum), with a weak peak of gaseous hydrogen. No dissolved hydrogen was detected in inclusions containing a water phase at higher pressures than 2 GPa. Occasionally, metastable water inclusions synthetized in the immiscibility region at pressures < 2.5 GPa were observed to nucleate a gas phase, triggered by laser ablation of nearby inclusions or upon laser irradiation during Raman analysis. In general, the density of water is relatively constant along the immiscibility curve from 1.5 to 3 GPa (Pitzer and Sterner 1994); it ranges from 1.02 g/cm3 at 1.5 GPa and 700 °C to 1.07 g/cm3 at 3 GPa and 1200 °C (the highest temperatures at which immiscibility is observed at a given pressures).
Inclusions where H2 gas is the dominant or only detectable phase are typically larger, compared to the size of mixed and pure water inclusions. According to Raman measurements, they appear to be either completely free from H2O or they contain only a thin coating of water along the walls of the inclusion. They also tend to be more resistant to recrystallization in our experiments, due to the extremely low solubility of major rock forming elements in non-polar hydrogen gas (Walther and Orville 1983). Combined with their size, this makes H2-rich inclusions easily detectable even at temperatures > 1000 °C (e.g. Figure 4c, e, f) and even in failed experiments, where water-containing inclusions show strong recrystallization features, such as negative crystal shapes or visible signs of fluid loss. Thus, a disappearance of distinctive hydrogen inclusions is a very reliable optical marker of full miscibility between H2O and H2. The disappearance of pure water inclusions is less noticeable, as they commonly exhibit vapour bubbles that may be confused with a hydrogen-rich gas phase without Raman analysis.

Synthetic fluid inclusions formed at 2.5–4 GPa in the immiscibility region, below the H2-H2O critical curve a, c, e, f and above it in the miscibility region b, d. Images c, d and f are combinations of several separate photographs taken at different focal depths
Fluids trapped in the inclusions contained some silica and magnesium, from the dissolution of quartz and forsterite. As water is diluted by non-polar hydrogen molecules, the solubility of silica decreases dramatically (Walther and Orville 1983). In laser-heated diamond cell experiments, the formation of SiH4 was observed as a result of the interaction of H2 and SiO2 (Shinozaki et al. 2014), however no detectable Raman signal of SiH4 was observed in our samples in the region of 2180–2300 cm−1. This suggests that this species either was not present in our experiments, or its concentration is negligible even in the low-density hydrogen-rich phase.
Evidence for a reduced melting temperature of iron in the presence of H2
In some of our experiments, we noted the occurrence of perfectly rounded droplets of metallic iron as inclusions in olivine crystals. This is consistent with the suggestion of Fukai et al. (2003) that alloying with H2 may strongly reduce the melting temperature of iron. As this observation is somewhat outside of the scope of this paper, the corresponding data are compiled in Figures S4 and S5 of the supplementary online material (file 2).
H2-H2O element partitioning experiments
Six experiments were performed (Table 2) in an attempt to investigate the partitioning of various elements between the coexisting H2-rich and H2O-rich fluid phases. Initially, elements capable of producing gaseous hydrides at our experimental conditions such as C, P, As, Se, and Te (Greenwood and Earnshaw 2012) were the primary targets of our investigation, as well as Xe, Cs, and Rb, which were planned to be used as internal standards. However, most of those experiments failed due to the reactivity of the selected chemical elements. Quantification of phosphorus in fluid inclusions in quartz (KVHP6) with the use of LA ICP-MS turned out to be impossible due to the incorporation of this element into newly-formed quartz, with the signal intensity from inclusion-free areas being comparable to the signal intensity from the inclusion itself. As, Se, and Te (KVHP7, KVHP8) were either completely lost to the inner platinum capsule, or they damaged the capsule during experiment due to the formation of platinum compounds. However, the experiments with C, Xe, Rb, Cs and other elements present in the doped diopside glass (KVHP9, KVHP10, KVHP11) were successful. Experiments KVHP9 and KVHP10 were carried out at 2 GPa and 750 °C, less than 100 ˚C below the critical curve. Though the inclusions are relatively small, the immiscibility can still be easily observed. Samples from the KVHP9 (supplementary file 2, Figure S2) experiment were analysed with LA ICP-MS, but the small size of the inclusions (~ 10–30 μm) did not allow to detect a clear fractionation effect between the water and hydrogen-rich phases. Inclusions were also produced in the experiment KVHP11 at 2.3 GPa and 675 °C (supplementary file 2, Figure S3), ~ 275 degrees below the critical curve. At those P–T conditions, inclusions were trapped deep in the immiscibility region to enhance any possible element partitioning. They range in size from 10 μm to 100 μm with multiple water and hydrogen inclusions of comparable size, while some inclusions with H2 are considerably larger (supplementary file 2, Figure S3b). Raman measurements of all types of inclusions and phases present in this sample yielded the first valuable result–hydrogen inclusions are rich in CH4 (supplementary file 2, Figure S3c, orange), while this species is almost absent from water inclusions. Only a minor CH4 signal can be seen in the vapour bubble of water-rich inclusions (supplementary file 2, Figure S3c, green and blue). Initially, carbon was added to the capsule in the form of CaCO3 as a minor impurity in the utilized Ca(OH)2. During the experiment in the Fe-FeO buffered capsule the presence of free hydrogen likely led to a rapid conversion of CO32− to CH4–a dominant carbon species at such conditions (C. Zhang and Duan 2009).
Further partitioning effects were unraveled with the use of LA ICP-MS with a typical inclusion laser ablation profile presented in Fig. 5 (full data available in supplementary file 1, Tables S1-S7). During ablation the gas phase is released first, hence the strongest xenon peak occurs at 32 s, followed by the remaining contents of the inclusion. As the fraction of gas phase in individual fluid inclusions increases (Fig. 6a) the Xe/Rb and Xe/Cs ratios tend to increase, demonstrating strong partitioning of alkali metals into the water phase. Xe, on the other hand, tends to partition into the gas phase. Although in similar-sized H2-rich versus H2O-rich fluid inclusions the intensities of the xenon signals are comparable (Fig. 6b), the lower density of the H2-rich phase implies strong partitioning of Xe into the H2 phase. At 2.3 GPa and 675 °C, the density of pure water is 1.138 g/cm3 (Wagner and Pruß 2002), and that of hydrogen 0.148 g/cm3 (Leachman et al. 2009). Accordingly, if for H2O-rich and H2-rich inclusions of similar size the intensity of the xenon signal is about the same, then the concentration of Xe (i.e., the weight of Xe relative to that of the bulk fluid) is about 8 times higher in the H2-rich phase, and Xe thus partitions by definition by a factor of about 8 into that phase (supplementary file 1, Table S1). The density of CH4, that is abundant in gas inclusions, at given experimental conditions is 0.58 g/cm3 (Jiang et al. 2022) which possibly reduces the partitioning coefficient, yet not by much since even pure methane is half as dense as water. The other trace elements measured, including the rare earths, alkalis, alkaline earths and various high-field strength elements, appear to partition strongly into the H2O-rich phase.

Laser ablation analysis of an individual gas-rich inclusion produced in the element partitioning experiment KVHP11

a LA-ICP-MS-established Xe/Cs count ratios plotted versus Xe/Rb count ratios for individual inclusions, and b 131Xe signal intensities plotted versus inclusion volume. Data for hydrogen-rich inclusions are shown in orange, whereas data for inclusions with liquid water and possible co-trapped hydrogen are shown in blue
Discussion
Comparison with previous data
The experimental method used in the current study to establish the position of the H2-H2O critical curve is very similar to the one of Bali et al. (2013). The only significant difference is the shorter run durations at temperatures above 850 °C (Table 1). As may be seen from Fig. 2, the experimental data are extremely similar in the pressure range where the two studies overlap, with both critical curves having the same slope. The only significant deviation between the two datasets that cannot be explained by thermocouple uncertainty or temperature gradients in the piston-cylinder assembly is a single experiment of Bali et al. at 2.3 GPa and 900 °C, showing immiscibility where our data suggests that fluids are still miscible. Yet in their work the critical curve was drawn not below this experimental point, but across it, which indicates that it may have been a borderline case located extremely close to the curve, where distinguishing between immiscibility or miscibility patterns is not straightforward. Additionally, the critical curve of Bali et al. exhibits a gradual change in slope as quartz is replaced by forsterite as host crystal. This effect was not well manifested in our data with the slope being in fact almost linear and continuous from 1.5 to 2.5 GPa in the range where the host crystal changed. Potentially such effects could result from hydrogen diffusion, affecting the inclusion composition and data interpretation. Hydrogen diffusion has been known to be quite rapid in quartz with significant shift in phase assemblage occurring at 700 °C in experiments lasting for 7 days (Mavrogenes and Bodnar, 1993). Yet this process is unlikely to be relevant for our experiments, since the inclusions are in equilibrium with the fluid during the run, while decompression and sample processing times have been minimized. Even though some minor hydrogen loss is possible, the Raman detection limit for H2 is as low as 1–3 bar (Mavrogenes and Bodnar 1994). This suggests that even in case of partial loss the inclusion type would be determined correctly.
Both in the experiments of Bali et al. (2013) and in those of the present study, fluid composition was controlled by an external Fe-FeO-H2O buffer. This buffer will produce H2 by the reaction Fe + H2O = FeO + H2; the H2 will diffuse into the inner capsule until the equilibrium H2/H2O ratio in the charge is reached. According to the observations from synthetic fluid inclusions in the range of full miscibility (e.g. Figure 4b), this fluid contains H2 and H2O in a molar ratio close to unity, consistent with thermodynamic predictions. However, due to the experimental setup, the H2/H2O ratio cannot be independently varied in these runs. It is therefore not impossible that for a somewhat different H2/H2O ratio, the miscibility gap could close at a somewhat higher temperature. Our experimental observations suggest, however, that this is unlikely to be a significant effect. The fluid inclusions (e.g. Figure 4c) show that at temperatures not far below our critical curve, nearly pure H2 coexists with nearly pure H2O. This implies that the temperature of complete miscibility is rather insensitive to the actual H2/H2O ratio.
The results of Seward and Franck (1981), who measured immiscibility in systems with variable H2-H2O fractions to 440 °C and 250 MPa, are located outside of our experimental range and just slightly below the extrapolation of our critical curve. Though the discrepancy between this extrapolation and Seward and Franck’s data is minor, the explanation for it is straightforward. Our 600 °C and 1 GPa experiment is probably located almost exactly on the critical curve itself, as the pure hydrogen inclusions in this run are small and comparatively rare. Thus, at 1 GPa the real slope may be slightly steeper than we assumed. The extrapolation would then almost perfectly align with the critical curve of Seward and Franck (1981).
A direct comparison of our experiments with the computational results of Bergermann et al. (2021), which support the existence of immiscibility between water and hydrogen, or with those of Soubiran and Militzer (2015), which suggest the absence of such an effect, is complicated. Due to the sensitivity of the simulations to the choice of the cutoff radius value and the imperfections of analytical two-body interaction potentials, the disagreement of existing computational studies with our experimental data is quite significant. The results of Bergermann et al. (2021) indicate that at temperatures above 1100 °C, the experimental critical curve approaches the simulated curve and intersects it at ~ 1350 °C. With no miscibility observed in our experiments above 3 GPa the experimental data for 3.5 and 4 GPa is in seeming agreement with the simulated critical curve, however the general difference in slopes of the two curves suggests that this agreement is partially coincidental.
H2-H2O immiscibility in Earth’s interior and the formation of super-reduced mineral assemblages
Several mechanisms for the formation of super-reduced mineral associations have been proposed in the past. A first hypothesis suggested a deep origin (e.g. R. Zhang et al. 2016), assuming that phases such as moissanite, native silicon, iron, nitrides and phosphides crystallized at depths exceeding 200 km, where oxygen fugacity is expected to be low, although by far not low enough to stabilize SiC (Frost and McCammon 2008). For example, the SiC grains coexisting with diamond, native Fe and FeO from the Luobusa ophiolites (Tibet) were assumed to have formed at pressures of 8.5–9 GPa (R. Zhang et al. 2016), corresponding to a depth of ~ 300 km.
A second hypothesis, described in detail by Bali et al. (2013), proposes that oxygen fugacities at the level of the IW buffer and below do not necessarily require deep mantle conditions. The authors suggest that low fugacities can be achieved locally, via gradual water removal from immiscible H2-H2O fluids even at relatively shallow conditions in the upper mantle. At first, reducing fluids may form at 150–200 km depth (Fig. 7a, b) via slab dehydration (Kawamoto 2018). As water and hydrogen fluids percolate through the mantle, water is being sequestered in hydrous melts (Ni et al. 2011; Shen and Keppler 1995), incorporated into nominally anhydrous minerals (Fei and Katsura 2020; Hirschmann et al. 2005; Mierdel et al. 2007) or hydrous phases (Smyth 2018). As the amount of available H2O in the system decreases, the oxygen fugacity decreases as well (Bali et al. 2013) making the formation of super reducing phases plausible. Golubkova et al. (2016) suggested a similar model of H2O removal from reducing fluids as a mechanism for attaining ultra-reducing conditions in the shallow mantle, although without explicitly referring to H2-H2O immiscibility.

a Archean (grey) (Mareschal and Jaupart 2006) and modern (orange) geotherms (Hasterok and Chapman 2011; Rudnick and Nyblade 1999), compared with the H2-H2O immiscibility curve. b Oxygen fugacity range of cratonic peridotites based on xenolith data (Creighton et al. 2009; Goncharov et al. 2012; Lazarov et al. 2009; McCammon and Kopylova 2004; ; Woodland and Koch 2003; Yaxley et al. 2012). The dashed blue line shows IW buffer conditions (Campbell et al. 2009) calculated along the hot Kalahari geotherm (Rudnick and Nyblade 1999), which is shown in a as the upper bound of the modern cratonic geotherms
Carbon-rich phases–graphite, diamond, or hydrocarbons–are extremely important parts of reduced environments in the upper mantle (Frost and McCammon 2008). Although a detailed investigation of carbon geochemistry in the presence of H2-H2O fluids at IW buffer conditions is beyond the scope of our study, we did observe significant methane generation from minor carbonate impurities (< 0.5 wt. %) in the capsule. Existing experiments investigating immiscibility in the C-H–O system (Li 2016) are in full agreement with our experiments and indicate that the presence of carbon at least does not move the critical curve to lower temperatures at pressures below 3 GPa. In general, if methane is available at upper mantle conditions, diamond may form during the evolution of such fluids in the lithospheric mantle (e.g. Matjuschkin et al. 2019; Thomassot et al. 2007). Additionally, metastable low-pressure, low-temperature diamonds have been found to form in CH4-rich fluid inclusions during the serpentinization of oceanic lithosphere (Pujol-Solà et al. 2020). However, it is currently unknown how widespread this process is. In general, metasomatic processes caused by CH4-H2-rich fluids have been well documented (Vitale Brovarone et al. 2017) in ophicarbonates from the Italian Alps with abundant graphite and fluid inclusions being present in the rocks. Observations from our element partitioning experiments suggest that within the of H2-H2O immiscibility field, methane partitions into the highly mobile H2-rich fluid phase that in turn may be a precursor for the formation of diamonds, graphite, or moissanite.
Native iron, nickel, iron silicide, platinum and other metals and alloys have been described in metal-enriched xenoliths from Avacha volcano that brought up samples from the mantle wedge below the Kamchatka peninsula (Ishimaru et al. 2009; Kepezhinskas et al. 2022). Formation conditions for this suite of rocks and associated metals are estimated as 792–1100 °C and 1–3 GPa (Kepezhinskas et al. 2022 and references within), consistent with the origin in a supra-subduction zone environment at rather shallow depth in the mantle, and can occur via the mechanism proposed by Bali et al. (2013) and in our study. In summary, our data suggest that super-reduced phases are not necessarily associated with UHP or UHT conditions, but can form at ~ 100–150 km or even shallower levels in the upper mantle through reactions involving reducing immiscible H2-CH4 fluids.
H2-H2O unmixing as a possible cause for mantle oxidation and the early loss of Earth’s noble gases
Isotopes of noble gases, especially of xenon and neon, have traditionally been used to constrain Earth’s early outgassing and atmosphere composition (e.g. Allègre et al. 1987; Marty 1989). The first atmospheres of Earth, Mars and Venus supposedly formed as the result of rapid gas release from the planetary interior, or by equilibration with a magma ocean, occurring shortly after core formation (Gaillard et al. 2022; Lammer et al. 2018; Trieloff et al. 2000). During those times the Earth’s upper mantle, according to the model of Rubie et al. (2011), was hotter than in the Archaean (Fig. 7a) and was extremely reduced. However, this state lasted only for a short time: Cerium concentrations in the oldest surviving zircons suggest rapid upper mantle oxidation (Smythe and Brenan 2016; Trail et al. 2011) that was completed 200 Myr after planetary formation, though the atmosphere itself may have remained reducing for a longer time (e.g. Kasting et al. 1993; Holland 2002).
Bali et al. (2013) suggested that rapid upper mantle oxidation was caused by the appearance of a miscibility gap between water and hydrogen when the planet cooled down. As noted by Sharp et al. (2013), rapid hydrogen degassing may also explain differences in water content and isotope compositions (H, Cl) in the Earth-Moon system. Hydrogen is easily transferred to the fluid phase during degassing of silicate melts (Hirschmann et al. 2012; Moore et al. 1998), thus the emergence of immiscibility between H2 and H2O may trigger the formation of a buoyant and low-viscosity H2-dominant phase, followed by its rapid upward percolation (Bali et al. 2013; Sharp et al. 2013). Such a scenario is plausible if geotherms crossed the immiscibility curve, intensifying hydrogen phase loss rate and driving the upper mantle oxygen fugacity to higher values. Our new data show a steep increase of the critical curve at pressures above 2 GPa (Fig. 7a). This suggests that even for very hot geotherms in the early Archean or late Hadean, H2-H2O immiscibility occurred in the deeper parts of the upper mantle. Therefore, the scenario of early mantle oxidation by selective loss of a highly buoyant H2-rich fluid phase is very plausible.
Noble gas isotope data supports this idea, suggesting increased outgassing flux during the first 20–30 Myr after core formation (Kunz et al. 1998). One of the anomalies observed in the Earth’s upper mantle noble gas compositions is the relative underabundance of 129Xe, produced via the decay of 129I (t1/2 = 1.6 × 107 yr) that went extinct ~ 100 Myr after the formation of the solar system (Mukhopadhyay 2012). Similarly, there is a striking deficiency of xenon isotopes produced by spontaneous fission of now-extinct Pu (Kunz et al. 1998). Xenon is the least soluble in silicate melts of all noble gases (Schmidt and Keppler 2002; Shibata et al. 1998) and thus one would expect it to preferentially partition into a fluid phase. As can be seen from our partitioning experiments, xenon enters preferentially the reduced H2-rich phase. Thus, the early and rapid decrease in radiogenic xenon isotopes in the mantle can be explained by its partitioning into a hydrogen-rich phase during unmixing, subsequent degassing and then loss from the Earth’s atmosphere for example by impact erosion during the late veneer event (Kruijer et al. 2015; Walker 2009).
Based on existing thermal evolution models (Herzberg et al. 2010; Korenaga 2006; Rubie et al. 2011) and degassing constraints (Kunz et al. 1998; Mukhopadhyay 2012), we estimate that H2-H2O unmixing may have occurred ~ 50–150 Myr after planetary formation, which is consistent with the evidences for an oxidized upper mantle 4350 Myr before present (Smythe and Brenan 2016; Trail et al. 2011). H2 degassing may not be exclusive to Earth but may have occurred on Mars and Venus as well. This makes rapid early hydrogen escape, related to H2-H2O immiscibility, a possible common feature of the evolution of rocky planets, with the underabundance of radiogenic 129Xe being a prominent marker of this process.
Conclusions
-
1.
The strong increase in the critical curve of the H2-H2O system observed above 2 GPa in this study implies that immiscibility between H2 and H2O may have occurred in the deeper parts of the upper mantle already very early in Earth´s history, when ambient mantle temperatures were much higher than today.
-
2.
We present the first experimental evidence that Xe–and by inference also other noble gases–strongly partitions into the H2-rich gas phase.
-
3.
Degassing of a H2-rich fluid from the mantle is a plausible mechanism for both the early oxidation of the mantle and for the selective loss of noble gases in the Hadean.
-
4.
In the modern Earth, the local occurrence of immiscible H2-rich fluids may be responsible for the formation of ultra-reduced mantle domains containing moissanite. These domains may form already at rather shallow depth in the mantle.
Data availability
All of the data supporting the findings of this study are included within the paper itself and its supplementary files.
References
Abbott D, Burgess L, Longhi J, Smith WHF (1994) An empirical thermal history of the Earth’s upper mantle. J Geophys Res 99(B7):13835–13850. https://doi.org/10.1029/94jb00112
Allègre CJ, Staudacher T, Sarda P (1987) Rare gas systematics: formation of the atmosphere, evolution and structure of the Earth’s mantle. Earth Planet Sci Lett 81(2–3):127–150. https://doi.org/10.1016/0012-821X(87)90151-8
Armstrong K, Frost D, McCammon C, Rubie D, Ballaran T (2019) Deep magma ocean formation set the oxidation state of Earth’s mantle. Science 365(6456):903–906. https://doi.org/10.1126/science.aax8376
Bali E, Audétat A, Keppler H (2013) Water and hydrogen are immiscible in Earth’s mantle. Nature 495(7440):220–222. https://doi.org/10.1038/nature11908
Ballhaus C (1993) Redox states of lithospheric and asthenospheric upper mantle. Contrib to Mineral Petrol 114(3):331–348. https://doi.org/10.1007/BF01046536
Bergermann A, French M, Redmer R (2021) Gibbs-ensemble monte carlo simulation of H2–H2O mixtures. Phys Chem Chem Phys 23(22):12637–12643. https://doi.org/10.1039/d1cp00515d
Bose K, Ganguly J (1995) Quartz-coesite transition revisited: reversed experimental determination at 500–1200 °C and retrieved thermochemical properties. Am Mineral 80(3–4):231–238. https://doi.org/10.2138/am-1995-3-404
Campbell A, Danielson L, Righter K, Seagle CT, Wang Y, Prakapenka V (2009) High pressure effects on the iron-iron oxide and nickel-nickel oxide oxygen fugacity buffers. Earth Planet Sci Lett 286(3–4):556–564. https://doi.org/10.1016/j.epsl.2009.07.022
Chou I (1987) Oxygen buffer and hydrogen sensor techniques at elevated pressures and temperatures. In: Ulmer G, Barnes H (eds) Hydrothermal Experimental Techniques. John Wiley, New York, pp 61–99
Creighton S, Stachel T, Matveev S, Höfer H, McCammon C, Luth RW (2009) Oxidation of the kaapvaal lithospheric mantle driven by metasomatism. Contrib to Min Petrol 157(4):491–504. https://doi.org/10.1007/s00410-008-0348-3
Dobrzhinetskaya L, Mukhin P, Wang Q, Wirth R, O’Bannon E, Zhao W, Eppelbaum L, Sokhonchuk T (2018) Moissanite (SiC) with metal-silicide and silicon inclusions from tuff of Israel: Raman spectroscopy and electron microscope studies. Lithos 310–311:355–368. https://doi.org/10.1016/j.lithos.2017.04.001
Fei H, Katsura T (2020) Pressure dependence of proton incorporation and water solubility in olivine. J Geophys Res Solid Earth 125(2):018813. https://doi.org/10.1029/2019JB018813
Frost D, McCammon C (2008) The redox state of Earth’s mantle. Annu Rev Earth Planet Sci 36(1):389–420. https://doi.org/10.1146/annurev.earth.36.031207.124322
Fukai Y, Mori K, Shinomiya H (2003) The phase diagram and superabundant vacancy formation in Fe-H alloys under high hydrogen pressures. J Alloys Compd 348(1–2):105–109. https://doi.org/10.1016/S0925-8388(02)00806-X
Gaillard F, Bernadou F, Roskosz M, Bouhifd MA, Marrocchi Y, Iacono-Marziano G, Moreira M, Scaillet B, Rogerie G (2022) Redox controls during magma ocean degassing. Earth Planet Sci Lett 577:117255
Golubkova A, Schmidt M, Connolly JAD (2016) Ultra-reducing conditions in average mantle peridotites and in podiform chromitites: a thermodynamic model for moissanite (SiC) formation. Contrib to Min Petrol 171(5):1–17
Goncharov A, Ionov D, Doucet L, Pokhilenko L (2012) Thermal state, oxygen fugacity and C-O-H fluid speciation in cratonic lithospheric mantle: New data on peridotite xenoliths from the Udachnaya kimberlite, Siberia. Earth Planet Sci Lett 357–358:99–110. https://doi.org/10.1016/j.epsl.2012.09.016
Greenwood N, Earnshaw A (2012) Chemistry of the Elements. Elsevier, Oxford
Hasterok D, Chapman D (2011) Heat production and geotherms for the continental lithosphere. Earth Planet Sci Lett 307(1–2):59–70. https://doi.org/10.1016/j.epsl.2011.04.034
Herzberg C, Condie K, Korenaga J (2010) Thermal history of the Earth and its petrological expression. Earth Planet Sci Lett 292(1–2):79–88. https://doi.org/10.1016/j.epsl.2010.01.022
Hirschmann M, Aubaud C, Withers A (2005) Storage capacity of H2O in nominally anhydrous minerals in the upper mantle. Earth Planet Sci Lett 236(1–2):167–181. https://doi.org/10.1016/j.epsl.2005.04.022
Hirschmann M, Withers A, Ardia P, Foley N (2012) Solubility of molecular hydrogen in silicate melts and consequences for volatile evolution of terrestrial planets. Earth Planet Sci Lett 345–348:38–48. https://doi.org/10.1016/j.epsl.2012.06.031
Holland H (2002) Volcanic gases, black smokers, and the great oxidation event. Geochim Cosmochim Acta 66(21):3811–3826. https://doi.org/10.1016/S0016-7037(02)00950-X
Ishimaru S, Arai S, Shukuno H (2009) Metal-saturated peridotite in the mantle wedge inferred from metal-bearing peridotite xenoliths from Avacha volcano. Kamchatka Earth Planet Sci Lett 284(3–4):352–360. https://doi.org/10.1016/j.epsl.2009.04.042
Jiang S, Guo T, Yu Y, Hu J (2022) Molecular dynamics simulation and a cubic equation of state of supercritical methane Up to 3000 K and 3000 MPa. Int J Thermophys 43:22. https://doi.org/10.1007/s10765-021-02952-4
Kaminsky F (2012) Mineralogy of the lower mantle: a review of “super-deep” mineral inclusions in diamond. Earth Sci Rev 110(1–4):127–147. https://doi.org/10.1016/j.earscirev.2011.10.005
Kasting J, Eggler D, Raeburn S (1993) Mantle redox evolution and the oxidation state of the Archean atmosphere. J Geol 101(2):245–257. https://doi.org/10.1086/648219
Kawamoto T (2018) Hydrous phases and water transport in the subducting slab. Rev Mineral Geochem 62:273–290. https://doi.org/10.2138/rmg.2006.62.12
Kepezhinskas P, Berdnikov N, Kepezhinskas N, Konovalova N (2022) Metals in Avachinsky peridotite xenoliths with implications for redox heterogeneity and metal enrichment in the Kamchatka mantle wedge. Lithos 412:106610. https://doi.org/10.1016/j.lithos.2022.106610
Korenaga J (2006) Archean geodynamics and the thermal evolution of Earth. Geophys Monogr Ser 164:7. https://doi.org/10.1029/164GM03
Kruijer T, Kleine T, Fischer-Gödde M, Sprung P (2015) Lunar tungsten isotopic evidence for the late veneer. Nature 520(7548):534–537. https://doi.org/10.1038/nature14360
Kunz J, Staudacher T, Allègre CJ (1998) Plutonium-fission xenon found in Earth’s mantle. Science 280(5365):877–880. https://doi.org/10.1126/science.280.5365.877
Lammer H, Zerkle AL, Gebauer S, Tosi N, Noack L, Scherf M, Pilat-Lohinger E, Güdel M, Grenfell JL, Godolt M, Nikolaou A (2018) Origin and evolution of the atmospheres of early Venus, Earth and Mars. Astron Astrophys Rev 26(1):1–72. https://doi.org/10.1007/s00159-018-0108-y
Lathe C, Koch-Müller M, Wirth R, van Westrenen W, Mueller H, Schilling F, Lauterjung J (2005) The influence of OH in coesite on the kinetics of the coesite-quartz phase transition. Am Miner 90(1):36–43. https://doi.org/10.2138/am.2005.1662
Lazarov M, Woodland A, Brey G (2009) Thermal state and redox conditions of the Kaapvaal mantle: A study of xenoliths from the Finsch mine, South Africa. Lithos 112:913–923. https://doi.org/10.1016/j.lithos.2009.03.035
Leachman J, Jacobsen R, Penoncello S, Lemmon E (2009) Fundamental equations of state for parahydrogen, normal hydrogen, and orthohydrogen. J Phys Chem Ref Data 38(3):721–748. https://doi.org/10.1063/1.3160306
Li Y (2016) Immiscible C-H-O fluids formed at subduction zone conditions. Geochem Perspect Lett. https://doi.org/10.7185/geochemlet.1702
Mareschal J, Jaupart C (2006) Archean geodynamics and environments. Geophys Monogr Ser 164:61–73
Marty B (1989) Neon and xenon isotopes in MORB: implications for the earth-atmosphere evolution. Earth Planet Sci Lett 94(1–2):45–56. https://doi.org/10.1016/0012-821X(89)90082-4
Matjuschkin V, Woodland A, Yaxley G (2019) Methane-bearing fluids in the upper mantle: an experimental approach. Contrib to Mineral Petrol 174(1):1–14. https://doi.org/10.1007/s00410-018-1536-4
Mavrogenes J, Bodnar R (1994) Hydrogen movement into and out of fluid inclusions in quartz: Experimental evidence and geologic implications. Geochim Cosmochim Acta 58(1):141–148. https://doi.org/10.1016/0016-7037(94)90452-9
McCammon C, Kopylova M (2004) A redox profile of the slave mantle and oxygen fugacity control in the cratonic mantle. Contrib to Mineral Petrol 148(1):55–68. https://doi.org/10.1007/s00410-004-0583-1
Mierdel K, Keppler H, Smyth J, Langenhorst F (2007) Water solubility in aluminous orthopyroxene and the origin of Earth’s asthenosphere. Science 315(5810):364–368. https://doi.org/10.1126/science.1135422
Moore G, Vennemann T, Carmichael I (1998) An empirical model for the solubility of H2O in magmas to 3 kilobars. Am Mineral 83(1–2):36–42. https://doi.org/10.2138/am-1998-1-203
Mukhopadhyay S (2012) Early differentiation and volatile accretion recorded in deep-mantle neon and xenon. Nature 486(7401):101–104. https://doi.org/10.1038/nature11141
Nazzareni S, Nestola F, Zanon V, Bindi L, Scricciolo E, Petrelli M, Zanatta M, Mariotto G, Giuli G (2019) Discovery of moissanite in a peralkaline syenite from the Azores Islands. Lithos 324–325:68–73. https://doi.org/10.1016/j.lithos.2018.10.036
Ni H, Keppler H, Behrens H (2011) Electrical conductivity of hydrous basaltic melts: implications for partial melting in the upper mantle. Contrib to Mineral Petrol 162(3):637–650. https://doi.org/10.1007/s00410-011-0617-4
Pitzer K, Sterner S (1994) Equations of state valid continuously from zero to extreme pressures for H2O and CO2. J Chem Phys 101(4):3111–3116. https://doi.org/10.1063/1.467624
Pujol-Solà N, Proenza JA, Garcia-Casco A, González-Jiménez JM, Andreazini A, Melgarejo JC, Gervilla F (2018) An alternative scenario on the origin of ultra-high pressure (UHP) and super-reduced (SuR) minerals in ophiolitic chromitites: A case study from the Mercedita deposit (Eastern Cuba). Minerals 8(10):433. https://doi.org/10.3390/min8100433
Pujol-Solà N, Garcia-Casco A, Proenza JA, González-Jiménez JM, del Campo A, Colás V, Canalis A, Sánchez-Navas A, Roqué-Rosell J (2020) Diamond forms during low pressure serpentinisation of oceanic lithosphere. Geochem Perspect Lett 15:19–24. https://doi.org/10.7185/geochemlet.2029
Rubie D, Frost D, Mann U, Asahara Y, Nimmo F, Tsuno K, Kegler P, Holzheid A, Palme H (2011) Heterogeneous accretion, composition and core-mantle differentiation of the Earth. Earth Planet Sci Lett 301(1–2):31–42. https://doi.org/10.1016/j.epsl.2010.11.030
Rudnick R, Nyblade A (1999) The thickness and heat production of archean lithosphere: constraints from xenolith thermobarometry and surface heat flow. mantle petrology: field observations and high pressure experimentation: a tribute to francis R. Boyd. 6:3–12
Rustioni G, Audétat A, Keppler H (2019) Experimental evidence for fluid-induced melting in subduction zones. Geochem Perspect Lett 11:49–54. https://doi.org/10.7185/geochemlet.1925
Schmidt B, Keppler H (2002) Experimental evidence for high noble gas solubilities in silicate melts under mantle pressures. Earth Planet Sci Lett 195(3–4):277–290. https://doi.org/10.1016/S0012-821X(01)00584-2
Seward T, Franck E (1981) System hydrogen water up to 440 °C and 2500 bar pressure. Ber Bunsenges Phys Chem 85(1):2–7. https://doi.org/10.1002/bbpc.1981085010
Sharp Z, McCubbin F, Shearer C (2013) A hydrogen-based oxidation mechanism relevant to planetary formation. Earth Planet Sci Lett 380:88–97. https://doi.org/10.1016/j.epsl.2013.08.015
Shen A, Keppler H (1995) Infrared spectroscopy of hydrous silicate melts to 1000°C and 10 kbar: direct observation of H2O speciation in a diamond-anvil cell. Am Mineral 80(11–12):1335–1338. https://doi.org/10.2138/am-1995-11-1223
Shibata T, Takahashi E, Matsuda J (1998) Solubility of neon, argon, krypton, and xenon in binary and ternary silicate systems: a new view on noble gas solubility. Geochim Cosmochim Acta 62(7):1241–1253. https://doi.org/10.1016/S0016-7037(98)00046-5
Shinozaki A, Kagi H, Noguchi N, Hirai H, Ohfuji H, Okada T, Nakano S, Yagi T (2014) Formation of SiH4 and H2O by the dissolution of quartz in H2 fluid under high pressure and temperature. Am Mineral 99(7):1265–1269. https://doi.org/10.2138/am.2014.4798
Shiryaev A, Griffin W, Stoyanov E (2011) Moissanite (SiC) from kimberlites: Polytypes, trace elements, inclusions and speculations on origin. Lithos 122(3–4):152–164. https://doi.org/10.1016/j.lithos.2010.12.011
Smyth J (2018) Hydrogen in high pressure silicate and oxide mineral structures. Rev Mineral Geochem 62(1):85–115. https://doi.org/10.2138/rmg.2006.62.5
Smythe D, Brenan J (2016) Magmatic oxygen fugacity estimated using zircon-melt partitioning of cerium. Earth Planet Sci Lett 453:260–266. https://doi.org/10.1016/j.epsl.2016.08.013
Soubiran F, Militzer B (2015) Miscibility calculations for water and hydrogen in giant planets. Astrophys J 806(2):228. https://doi.org/10.1088/0004-637X/806/2/228
Thomassot E, Cartigny P, Harris J (2007) Methane-related diamond crystallization in the Earth’s mantle: Stable isotope evidences from a single diamond-bearing xenolith. Earth Planet Sci Lett 257(3–4):362–371. https://doi.org/10.1016/j.epsl.2007.02.020
Tingle T, Roedder E, Green H (1992) Formation of fluid inclusions and etch tunnels in olivine at high pressure. Am Mineral 77(3–4):296–302
Trail D, Watson E, Tailby N (2011) The oxidation state of Hadean magmas and implications for early Earth’s atmosphere. Nature 480(7375):79–82. https://doi.org/10.1038/nature10655
Trieloff M, Kunz J, Clague D, Harrison D, Allègre C (2000) The nature of pristine noble gases in mantle plumes. Science 288(5468):1036–1038. https://doi.org/10.1126/science.288.5468.1036
Trumbull R, Yang J-S, Robinson P, Di Pierro S, Vennemann T, Wiedenbeck M (2009) The carbon isotope composition of natural SiC (moissanite) from the Earth’s mantle: New discoveries from ophiolites. Lithos 113(3–4):612–620. https://doi.org/10.1016/j.lithos.2009.06.033
Tuff J, Wade J, Wood B (2013) Volcanism on Mars controlled by early oxidation of the upper mantle. Nature 498(7454):342–345. https://doi.org/10.1038/nature12225
Ulmer G, Grandstaff D, Woermann E, Göbbels M, Schönitz M, Woodland A (1998) The redox stability of moissanite (SiC) compared with metal-metal oxide buffers at 1773 K and at pressures up to 90 kbar. Neues Jahrb Für Mineral Abhandlungen 172(2–3):279–307. https://doi.org/10.1127/njma/172/1998/279
Vitale Brovarone A, Martinez I, Elmaleh A, Compagnoni R, Chaduteau C, Ferraris C, Esteve I (2017) Massive production of abiotic methane during subduction evidenced in metamorphosed ophicarbonates from the Italian Alps. Nat Commun 8:14134. https://doi.org/10.1038/ncomms14134
Wagner W, Pruß A (2002) The IAPWS formulation 1995 for the thermodynamic properties of ordinary water substance for general and scientific use. J Phys Chem Ref Data 31(2):387–535. https://doi.org/10.1063/1.1461829
Walker R (2009) Highly siderophile elements in the earth, moon and mars: update and implications for planetary accretion and differentiation. Chem Erde 69(2):101–125. https://doi.org/10.1016/j.chemer.2008.10.001
Walther J, Orville P (1983) The extraction-quench technique for determination of the thermodynamic properties of solute complexes: application to quartz solubility in fluid mixtures. Am Mineral 68(7–8):731–741
Williams H, Wood B, Wade J, Frost D, Tuff J (2012) Isotopic evidence for internal oxidation of the Earth’s mantle during accretion. Earth Planet Sci Lett 321–322:54–63. https://doi.org/10.1016/j.epsl.2011.12.030
Woodland A, Koch M (2003) Variation in oxygen fugacity with depth in the upper mantle beneath the Kaapvaal craton. Southern Africa Earth Planet Sci Lett 214(1–2):295–310. https://doi.org/10.1016/S0012-821X(03)00379-0
Wu S, Audétat A, Jochum K, Wang H, Chen J, Stoll B, Zhang C, Bao Z, Yang S, Li C, Wang X, Xu C, Xu L, Huang C, Xie L, Yang Y, Yang J (2022) three natural andesitic to rhyolitic glasses (OJY-1, OH-1, OA-1) as reference materials for in situ microanalysis. Geostand Geoanal Res. https://doi.org/10.1111/ggr.12449
Yang X, Keppler H, Li Y (2016) Molecular hydrogen in mantle minerals. Geochem Perspect Lett 2(2):160–168. https://doi.org/10.7185/geochemlet.1616
Yaxley G, Berry A, Kamenetsky V, Woodland A, Golovin A (2012) An oxygen fugacity profile through the Siberian Craton - Fe K-edge XANES determinations of Fe3+/∑ Fe in garnets in peridotite xenoliths from the Udachnaya East kimberlite. Lithos 140–141:142–151. https://doi.org/10.1016/j.lithos.2012.01.016
Zhang C, Duan Z (2009) A model for C-O-H fluid in the Earth’s mantle. Geochim Cosmochim Acta 73(7):2089–2102. https://doi.org/10.1016/j.gca.2009.01.021
Zhang R, Yang J-S, Ernst W, Jahn B, Iizuka Y, Guo G (2016) Discovery of in situ super-reducing, ultrahigh-pressure phases in the Luobusa ophiolitic chromitites, tibet: new insights into the deep upper mantle and mantle transition zone. Am Mineral 101(6):1285–1294. https://doi.org/10.2138/am-2016-5436
Acknowledgements
We would like to thank Detlef Krauße for SEM & WDS analyses, Raphael Njul and Alexander Rother for sample preparation and polishing, as well as Greta Rustioni for providing the glass sample MORB2-2D2. This study was partially supported by Deutsche Forschungsgemeinschaft through the international research Training Network “Deep Earth Volatile Cycles” GRK 2156/1. Constructive reviews by Dimitri Sverjensky and by two anonymous referees helped to improve the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Dante Canil.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vlasov, K., Audétat, A. & Keppler, H. H2-H2O immiscibility in Earth’s upper mantle. Contrib Mineral Petrol 178, 36 (2023). https://doi.org/10.1007/s00410-023-02019-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-023-02019-7