
Abstract
The paper reports studies of the effects of ester side-group size and structure on chain transfer to polymer (CTP) and mol% branches (%br) in the final polymers from monomer-starved semi-batch emulsion homopolymerizations of a series of alkyl acrylates with different numbers of ester OR side-group carbon atoms (NsgC) in the range 1–10. Although the high instantaneous conversions and high gel contents in the final polymers show that intermolecular CTP is significant, the kinetics have been analysed principally in terms of intramolecular CTP (intraCTP) because this is expected to be numerically the dominant CTP process. For linear OR side groups, there is a small, continuous increase in %br (from ~ 3.4 to ~ 5.0 mol%) as NsgC increases from 1 to 8, which arises predominantly from the acrylate molar mass (Macrylate) reducing the molar monomer concentration and from the small increase in rate coefficient for propagation (kp) as NsgC increases; no discernible effects of NsgC on the rate coefficient for intraCTP (\({k}_{\mathrm{trP}}^{\mathrm{intra}}\)) are evident. For isomeric butyl acrylates, there is no measurable effect of isomer structure on %br (4.3–4.4 mol%), indicating that butyl groups are too small for changes in side-group bulkiness to influence \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) and kp. A similar observation was made for n-hexyl acrylate (nHA) and 2-ethylbutyl acrylate (%br 4.7–4.8 mol%). However, cyclohexyl acrylate (cHA) gives a much higher %br (~ 7.2 mol%), which has been assigned to steric effects increasing \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) because kp values for nHA and cHA are very similar. For OR side-groups with NsgC of 8 and10, there is a strong effect of side-group non-linearity (2-ethylhexyl, iso-octyl and iso-decyl) increasing %br, with polymers from n-alkyl acrylates having much lower %br (~ 4.5–5.0 mol%) than those from the equivalent non-linear acrylates (~ 6.7–7.1 mol%); since the kp values are expected to be similar for these acrylates, it is evident that the much bulkier non-linear OR groups cause a significant increase in \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) compared to the linear equivalents. Tentative hypotheses for these steric effects have been postulated. Studies of the effect of temperature (0–80 °C) on CTP and %br for methyl acrylate solution homopolymerization (in which intraCTP should be completely dominant) show the expected reduction in %br as temperature reduces, such that CTP is negligible at 0 °C; Arrhenius analysis gives an activation energy of 40.7 kJ mol−1 for intraCTP, which is within the range of values reported for n-butyl acrylate.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chain transfer to polymer (CTP) in radical polymerizations of acrylates via H-abstraction from the backbone tertiary C–H bond (thereby forming a more stable tertiary radical) was recognised as being of potential significance for the polymerization kinetics [1,2,3] well before the first experimental evidence (from detection of the resulting branches using 13C NMR spectroscopy) was reported, not only of its existence, but also of its high prevalence in emulsion copolymerizations of n-butyl acrylate (nBA) with small proportions of acrylic acid and methyl methacrylate [4,5,6]. However, it was not until we reported studies of nBA solution homopolymerization [7] that the radical polymerization kinetics community recognised its significance in complicating accurate measurements of propagation rate coefficients by pulsed laser polymerization (PLP) [8,9,10,11]. This recognition catalysed a considerable amount of research into better understanding and overcoming the issues created by CTP [12, 13], an important development being the use of higher-frequency lasers to reduce or eliminate the contributions of CTP to the measurements [12,13,14,15,16].
Soon after the reports of CTP in nBA-based polymerizations, it was reported that CTP occurs in free-radical solution [17] and emulsion [18] homopolymerizations of 2-ethylhexyl acrylate (2EHA), an important principal monomer for production of water-borne pressure-sensitive adhesives. These observations confirmed that CTP is likely to be significant in radical polymerizations of all acrylate monomers. A further important observation was that CTP for 2EHA occurred to a much higher extent (typically about 1.5 × higher) than for nBA and that CTP proceeded in the usual manner by H-abstraction from the backbone C–H bond, i.e., there was no evidence for H-abstraction from the side-group tertiary C–H bond [17], which was rationalised in terms of the strong additional stabilisation of the product radical through resonance with the ester carbonyl group for H-abstraction from the backbone C–H bond. The much higher extent of CTP for 2EHA than for nBA showed that the ester OR side-group structure can influence CTP and prompted the studies reported here for CTP in free-radical homopolymerizations of a wide range of acrylates with different side-groups.
Experimental
Materials
Source and purity information for the acrylate monomers is given in Table S1. All monomers were treated to remove the inhibitor by washing three times with equal volumes of 2% (w/v) aqueous sodium hydroxide solution, followed by washing several times with equal volumes of deionised water before drying over anhydrous calcium chloride at 4 °C overnight. For solution polymerization studies, methyl acrylate was further purified by distilling the inhibitor-free monomer under nitrogen at reduced pressure, collecting a fraction that had > 99.8% purity (measured by gas chromatography). Methyl 2,2-dimethylpropionate (Lancaster, 99%) was distilled under nitrogen before use, the fraction collected having a purity > 99.8% (measured by gas chromatography). Benzoyl peroxide (Merck-Schuchardt, ~ 75%, supplied wetted with water) was recrystallised from chloroform by the addition of twice the volume of methanol and dried in a vacuum oven at 25–30 °C. Water was deionised by passage through a series of Milli-RO6 (Millipore) ion exchange columns. Potassium persulphate (Aldrich, 99 + %), sodium bicarbonate (Aldrich, 99%), sodium dodecyl sulphate (Aldrich, 98%), tetrahydrofuran (BDH, 99 + %) and N,N-dimethyl-p-toluidine (Aldrich, 99%) were used as supplied.
Emulsion homopolymerization of acrylates
All emulsion polymerizations were performed at 0.3–1 kg scale using the formulation given in Table 1 (which gives quantities for a 1 kg scale). The initial sodium dodecyl sulphate, sodium bicarbonate and water were weighed into a flanged reaction vessel which was then equipped with a nitrogen inlet, condenser and mechanical paddle stirrer before being placed in a water bath at 80 ± 1 °C. A steady flow of nitrogen was established with stirring and 30 min allowed for the surfactant/buffer solution to attain the bath temperature and become deoxygenated. The first-stage monomer was then added and 5 min allowed for the temperature to stabilise before adding the first-stage solution of potassium persulphate in water. The reaction was allowed to proceed for 60 min before beginning addition of the second-stage monomer and aqueous surfactant/initiator solution feeds, these being metered at constant rates over a period of 180 min using Watson-Marlow peristaltic pumps (or KD Scientific syringe pumps for reactions at 0.3 kg scale). On completion of the second-stage feeds, a further 60 min reaction time was allowed for completion of the polymerization before cooling the latex to room temperature and filtering it through a 53 μm sieve. The steady flow of nitrogen and stirring were maintained throughout the complete reaction period.
For each reaction, aliquots (~ 6 g) were removed at 30 min intervals throughout the reaction and quenched by transferring the aliquot directly to a glass vial that was placed in an ice/water bath and contained an accurately pre-weighed quantity (~ 0.5 g) of a 5.0 wt% aqueous hydroquinone solution. Conversion was determined from duplicate measurements of solids content using a mass-balance approach to account for previously removed aliquots and non-polymeric solids. The z-average particle diameter (dz) was measured using a Brookhaven light scattering instrument equipped with a 20 mW HeNe laser and a BI-9000AT digital correlator. Analyses were conducted at 25 ± 0.1 °C for a period of 60 s using a 90° scattering angle. The latex aliquot was diluted in a 12 mm diameter glass cell using deionised water that had been filtered through a 0.22 µm filter, and the concentration adjusted until the count rate at the detector was within the required limits of 50–150 kcounts s−1. A minimum of 30 separate measurements of dz were obtained for each diluted latex sample using the Brookhaven cumulants method of data analysis and the mean value of dz taken.
For the final latexes, gel fractions in the polymers were determined by Soxhlet extraction. A latex sample was dialyzed against a 1000 × excess of deionised water for 14 days, followed by isolation of the polymer by freeze–thaw cycling. The wet polymer coagulum was washed thoroughly with deionised water and then dried to constant mass at 50 °C in a vacuum oven. An accurately pre-weighed quantity of the dry polymer (~ 1.2–1.3 g) was extracted with tetrahydrofuran for 48 h in a microporous glass thimble, and the resulting gel was dried to constant mass at 50 °C in a vacuum oven.
Solution polymerization of methyl acrylate
Solution polymerizations of methyl acrylate were performed at 0, 20, 40, 60, 70 and 80 °C under a flowing nitrogen atmosphere. Methyl 2,2-dimethylpropionate (66.05 g), methyl acrylate (20.00 g; 0.232 mol) and N,N-dimethyl-p-toluidine (0.447 g; 3.31 mmol) were added to a 250 ml flanged reaction vessel equipped with a mechanical paddle stirrer, condenser and nitrogen inlet. To remove oxygen and allow the reactants to attain the polymerization temperature, the initial charge was stirred for 60 min for the reaction at 0 °C and for 30 min for the reactions at 20–80 °C. After this initial period, a solution of benzoyl peroxide (0.800 g; 3.30 mmol) in methyl 2,2-dimethylpropionate (12.70 g) was added at a constant rate over a period of 60 min using a KD Scientific syringe pump. Following complete addition of the initiator solution, the reactants were stirred for a further 30 min before warming or cooling the polymer solution to room temperature.
The conversions of monomer to polymer for the final polymer solutions were determined by measurement of solids content. The final polymer solution (~ 5 g) was weighed accurately into a pre-weighed aluminium pan and allowed to dry overnight at room temperature. The sample was then dried to constant weight in a vacuum oven at 30–35 °C. All analyses were performed in duplicate, and mean values taken.
The molar mass distribution (MMD) of each final polymer was determined by gel permeation chromatography (GPC) using samples isolated as described for the conversion measurements. Solutions (0.20%(w/v)) of each polymer were prepared in tetrahydrofuran and injected (200 μl) into a GPC system comprising an in-line series of three poly(styrene-co-divinylbenzene) packed columns (Phenomenex; 300 mm length; 7.8 mm internal diameter; 5 μm bead diameter) with bead porosities in the order 105, 103 and 50 nm, respectively. The flow rate of the tetrahydrofuran eluent was 1 ml min−1. The eluent was monitored using a differential refractometer and the data collected and analysed using Polymer Laboratories Caliber (version 6.0) GPC software. The GPC apparatus was calibrated using narrow MMD poly(methyl methacrylate) standards (Polymer Laboratories) in the range 0.62–1400 kg mol−1.
13C NMR spectroscopy
Samples of the final polymers for analysis by 13C NMR spectroscopy were isolated (i) as described for gel fraction measurements for the emulsion homopolymerizations and (ii) as described for the conversion measurements for the methyl acrylate solution homopolymerizations. The isolated polymers were either dissolved (solution homopolymerizations) or swollen (emulsion homopolymerizations) in CDCl3 to a concentration of ~ 100 mg ml−1. 13C NMR spectra were recorded at room temperature using a Varian Associates Unity 500 spectrometer operating at 125.8 MHz. Chemical shifts were referenced to the solvent resonance, which was taken to lie at δC = 77.0. Fast pulse spectra were recorded with continuous proton decoupling using a pulse interval of 0.5 s and a flip angle of 45°. Under these rapid pulsing conditions, the relative intensities do not necessarily reflect the relative abundance of each type of carbon because of differential relaxation times and nuclear Overhauser enhancements (NOE). In particular, the intensities of Cq carbons are underestimated relative to CH and CH2 carbons. Hence, the polymers also were analysed for extended periods (> 12 h) under quantitative conditions using a pulse interval of ≳ 5 s (mostly10 s) and suppression of NOE by inverse gated decoupling. All spectra were run at least twice. Assignment of peaks was done as described previously [4, 6, 7, 17] using already-established peak assignments, the distortionless enhancement by polarisation transfer (DEPT) technique and chemical shift predictions.
Results and discussion
CTP in acrylate radical polymerizations can proceed intermolecularly or intramolecularly (also referred to as backbiting) with consequences for branching and crosslinking in the polymers produced, as shown in Scheme 1. The driving force for CTP is creation of a more stable tertiary chain radical (of type B or type D) from the normal secondary propagating chain radical due to the usual steric effects of reducing radical reactivity and a doubling of the level of resonance stabilisation by hyperconjugation with the adjacent CH2 groups (both types of chain radical are stabilised equivalently by resonance with the ester carbonyl group). Fragmentation of the product radicals of type B and type D by β-scission also can occur and gives a linear chain radical plus a dead polymer chain with a terminal C = C bond (a macromonomer), which if it polymerises will give a propagating chain with a long-chain branch that is identical in branch structure to those resulting from intermolecular CTP. In our previous studies of 2EHA free-radical homopolymerization [17], as well as in the present work, there was no evidence for terminal C = C bonds in the 13C NMR spectra of the polymers produced, indicating that if β-scission did occur, most of the macromonomers generated were consumed in the polymerizations. In view of this, and because β-scission tends to become the dominant process only at high reaction temperatures (typically > > 100 °C) in acrylate polymerizations taken to high conversions [19], β-scission is not shown in Scheme 1. Nevertheless, it should be noted, that mass spectroscopic analyses of lower molar mass species in polymers from nBA [20] and 2EHA [21] bulk homopolymerizations have revealed that β-scission can make measurable contributions at temperatures of 60–100 °C.

Chemistry of (a) intermolecular CTP and (b) intramolecular CTP (backbiting) in acrylate homopolymerizations. Intermolecular CTP gives rise to long-chain branches and crosslinking. Intramolecular CTP proceeds via a 6-membered ring conformation at the chain end and gives rise to short branches comprising 4 backbone carbon atoms (i.e. 2 repeat units long) that are referred to as short-chain branches. Local structures (c) in the polymer chains can be detected by 13C NMR spectroscopy, which gives unique peaks for (i) normal backbone CH and CH2 carbons, (ii) backbone CH2 carbons in chain-end repeat units and (iii) branch Cq, CH and CH2 carbons
A simplified kinetics analysis [22], based on Scheme 1, leads to the following instantaneous equation for the mol% of branched repeat units (referred to as the mol% branches, %br):
where \({k}_{\mathrm{trP}}^{\mathrm{inter}}\), \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) and \({k}_{p}\) are the rate coefficients for intermolecular CTP, intramolecular CTP and propagation of normal secondary chain radicals, and [P] and [M] are the local concentrations of polymer repeat units and monomer in the proximity of the propagating chains (e.g., in emulsion polymerizations, they are the concentrations within the polymer particles). In terms of correlating data for %br determined by 13C NMR spectroscopy with extent of CTP, it is very important to recognise that this equation is based upon the assumption that every CTP event leads to a branch, i.e. that addition of monomer is the only fate for the tertiary chain radicals of type B and type D produced by CTP. The limitations of Eq. (1) arising from this assumption have been discussed thoroughly elsewhere [22]. For the discussions in the present paper, it is important to consider the effects of the parameters in Eq. (1). For both inter- and intra-molecular CTP, %br is dependent on the ratio of rate coefficients for CTP and propagation, and since the activation energy for CTP must be higher than for propagation, %br will increase as temperature increases. Intermolecular CTP also depends on [P]/[M] and so will make a larger contribution to %br as conversion increases and/or under conditions where [P]/[M] is high, such as in emulsion polymerizations where propagation takes place predominantly within polymer particles (which becomes especially significant when the reaction is operated under the usual starved conditions in which the particles contain ≤ 20%, and often < 5–10%, monomer). Intramolecular CTP depends on 1/[M] and so makes an increasing contribution to %br as [M] reduces and will be completely dominant in dilute solution polymerizations or at low conversions in bulk polymerizations because individual dead and propagating polymer chains are isolated from each other.
In studies of the synthesis of latexes designed as models for commercial nBA-based water-borne pressure-sensitive adhesives, we showed that the branches resulting from CTP could be detected by careful integration of 13C NMR spectra obtained from accumulation of many thousands of pulses [4,5,6]. Although it is not possible to differentiate between short- and long-chain branches by 13C NMR spectroscopy because the resulting branch points have identical local structure (see Scheme 1), the total level of branching could easily be determined and revealed high levels of branching (typically 2–4 mol% of repeat units) that are similar in magnitude to those in low-density polyethylene [23,24,25], which is considered to be a very highly branched polymer.
Mass spectroscopic analysis of the relatively low molar mass polymers produced in nitroxide-mediated homopolymerizations of nBA revealed that intramolecular CTP is the dominant kinetic event [26], an observation that is now widely accepted as being more general in radical polymerizations of acrylates and in accord with well-established knowledge of CTP in high-temperature (typically > 150 °C) radical polymerizations of ethylene [23,24,25]. Intramolecular CTP gives rise to short-chain branches that have a major effect on the properties of polyethylene because they disrupt crystallisation and greatly reduce its degree of crystallinity [23, 27]. In contrast, acrylate-based polymers produced by radical polymerization are atactic and amorphous, so the short-chain branches arising from intramolecular CTP have only a small effect on the polymer properties. Intermolecular CTP, although numerically much less frequent, is very important for acrylates because it gives rise to long-chain branches and crosslinking, both of which have major effects on the mechanical, relaxational and rheological properties of the polymers produced, effects that are particularly important for the performance of pressure-sensitive adhesives [28,29,30,31].
Effects of NOE and pulse interval on accuracy of mol% branches measurements for acrylate homopolymers
When comparing the extent of CTP for different monomers and reaction conditions, knowledge of the accuracy of the %br data is crucial. In this section, discussion is restricted to the effect of NMR conditions on the accuracy of %br values. Discussion of the effects of acrylate side-group structure on the extent of CTP is given in the subsequent sections.
For the following discussion, it is helpful to view Scheme 1c. All acrylate homopolymers give normal backbone CH peaks at δC 41–42 and normal backbone CH2 peaks at δC 34–36. In our original work, we established two methods for calculation of %br: (i) using the branch Cq peak, which is observed clearly at δC ~ 48 and (ii) using the peaks for the branch CH and CH2 carbons, which are observed at δC 39–40 and δC 36–38, respectively, the integrations for which needed to be done very carefully. We found that for spectra recorded using fast pulse sequences (0.5 s interval between pulses), the branch Cq gave low values of %br compared to values determined from spectra obtained with slow pulsing (10.5 s pulse interval) and NOE suppression (by inverse gated decoupling). However, within experimental error, the %br data determined from the branch CH and CH2 were the same when recorded under both NMR conditions, indicating that the relaxation times for the branch CH and CH2 (compared to the normal and chain-end backbone CH and CH2) were not significantly affected by their proximity to the branch point and that NOE suppression most probably was not important. Thus, use of the branch CH and CH2 peak integrations made it possible to obtain accurate measurements of %br much more efficiently in terms of NMR spectrometer time. Despite this, all %br data reported by other groups since our original papers [4,5,6,7, 17] have been calculated using the branch Cq peak, some under non-quantitative fast pulse conditions. In the present work, we have explored further the effects of pulse interval and NOE suppression on the measured values of %br. One or more methods of calculation have been used to determine %br from the 13C NMR spectra of the acrylate homopolymers, employing Eqs. (2)–(5); the complete set of %br results obtained under different NMR conditions is presented in Table S2.
where IbrCq is the integration for the branch Cq peak, \(I_{\mathrm {nbCH+CH}_{2}}\) is the integration for the normal backbone CH + CH2 peaks, \(I_{\mathrm {brCH+CH}_{2}}\) is the integration for the branch CH + CH2 peaks, IbrCH is the integration for the branch CH peak, \(I_{\mathrm {eCH}_{2}}\) is the integration for the chain-end backbone CH2 peak, \(I_{\mathrm {peCH}_{2}}\) is the integration for the penultimate chain-end backbone CH2 peak and \(I_{\mathrm {sgOCH}_{x}}\) is the integration for the side-group OCHx peak. For spectra where the penultimate chain-end backbone CH2 peak could not be resolved from side-group carbon peaks, \({I_{\mathrm {peCH}_{2}}}\) was replaced by \(I_{\mathrm {eCH}_{2}}\) (since each chain-end should have one each of these CH2 groups). For spectra where both the penultimate chain-end and chain-end backbone CH2 peaks could not be resolved from side-group carbon peaks, \(I_{\mathrm {eCH}_{2}} + I_{\mathrm {peCH}_{2}}\) was replaced by \(I_{\mathrm {brCH}}{_+}{_{\mathrm {CH}_{2}}}/3\) or \(2I{\mathrm {_{brCH}}}/3\) (since one chain end should result from each CTP event, given that CTP is prevalent in acrylate homopolymerizations).
Figure 1a shows the ratio of the %br value calculated from spectra recorded with a long pulse delay without NOE suppression to the mean %br determined from spectra recorded under quantitative conditions with NOE suppression (by gated decoupling) and a long pulse interval (≳ 5 s). It can be seen that, within experimental error (estimated to be ± 10% of the %br value), NOE suppression has a discernible effect only on values of %br when the O–CHx side-group carbon peak is used as the normalising factor for the total number of repeat units; the value of %br is then 75 ± 11% of the accurate value. For the calculations of %br using branch Cq and branch CH + CH2 peaks referenced to the total backbone carbon signal, i.e. from Eqs. (2), (3) and (4), the absence of NOE suppression causes, at most, a small reduction in %br giving values that, on average, are 94 ± 5% and 94 ± 9% of the accurate value.

Values for (a) the ratio of %br calculated from slow pulse 13C NMR spectra without NOE suppression to those calculated from spectra recorded under quantitative conditions and (b) the ratio of %br calculated from fast pulse 13C NMR spectra to those calculated from spectra recorded under quantitative conditions: open circle, br Cq referenced to half the total backbone carbons, calculated using Eq. (2); open triangle, br Cq referenced to the total O–CHx side-group carbons, calculated using Eq. (5); and filled circle, br CH + CH2 referenced to 3 × the total backbone carbons, calculated using Eq. (3) (for 2EHA, these data are for br CH referenced to 1.5 × the total backbone carbons, calculated using Eq. (4) — the reasons for this are given in the discussion of higher acrylates). The grey regions represent the experimental error in determination of %br from 13C NMR spectra for these polymers
Figure 1b shows the ratio of %br from fast pulse spectra to the mean mol% branches determined from spectra recorded under quantitative conditions with NOE suppression (by gated decoupling) and a long pulse interval (≳ 5 s). Use of the fast pulse branch CH and CH2 peaks referenced to the total backbone carbon signal, i.e. calculated using Eq. (3), gives values that are 98 ± 6% of the correct value, which is within the experimental error. Thus, as previously observed [4,5,6,7, 17], calculation of %br from the branch CH and CH2 peaks using Eq. (3) or (4) gives accurate values for spectra recorded under fast pulse conditions with no NOE suppression. However, use of the branch Cq peak to calculate %br from spectra recorded under fast pulse conditions gives values that, on average are 58 ± 4% of the correct value when referenced to the total backbone carbon signal, i.e. calculated using Eq. (2), and 46 ± 5% of the correct value when referenced to the O–CHx side-group carbon peak, i.e. calculated using Eq. (5). Hence, for the majority of acrylate homopolymers, there is a benefit from using branch CH and CH2 peaks with Eqs. (3) and (4) to determine %br because the amount of NMR time required to obtain reliably accurate %br data is greatly reduced (typically by a factor of 10–20 × compared to spectra recorded with long pulse intervals), especially given the typically high demand for NMR spectrometer time.
Since most of the polymers in our evaluation of the effects of ester side-group on the extent of CTP had not been studied previously, spectra were recorded using the quantitative conditions of NOE suppression and long pulse delays for the majority of the polymers — only %br data calculated from these spectra are presented for discussion of these polymers in the following sections. For the polymers analysed exclusively under fast pulse conditions (PMA, PEA and PiBA), only %br calculated from the branch CH + CH2 peaks using Eq. (3) are included in the discussion because, as shown above, they can be taken to be reliably accurate. Hence, all %br data discussed in the subsequent sections are considered accurate within the experimental error (approximately ± 10% of the value for %br ≥ 1 mol%).
Effects of side-group on the acrylate emulsion homopolymerizations
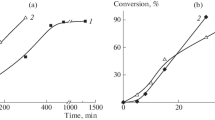
All the acrylate emulsion homopolymerizations were performed using the same formulation and procedure in which 7.5% of the monomer and 13% of the surfactant were used in a seed-stage to create the initial particles. The remaining monomer and surfactant were fed to the reactor in parallel over a 3 h period, which was followed by a final 1 h at reaction temperature to complete the polymerization. The scale of the reaction had to be reduced for monomers that could be obtained only in smaller quantities (see Table 2). With few exceptions, the emulsion polymerizations proceeded very similarly with high instantaneous conversions throughout the second-stage monomer feeding, very high final conversions, relatively low levels of coagulum, similar final particle diameters and high levels of crosslinked polymer (gel) in the final polymers. Representative plots for the evolution of conversion and particle diameter with reaction time are shown in Fig. 2 for two typical and one atypical reaction (plots are given for each monomer in Fig. S1).

Variation of overall conversion (open square), instantaneous conversion (open circle) and dz (filled circle) with reaction time for emulsion homopolymerization. The plots for nBA and iOA are representative of most of the monomers (MA, EA, nBA, iBA, tBA, nHA, 2EBA, cHA and iOA), whereas the plot for sBA is representative for monomers (sBA, nDA and iDA) with lower instantaneous conversions during the monomer feed
For most of the acrylates, polymerization of the majority of the monomer proceeded with high instantaneous conversions, typically > 80% (see Figs. 2 and S1), though for sBA, nDA and iDA, the instantaneous conversions were lower, but still > 60% for most of the monomer conversion. The high instantaneous conversions and high levels of gel in the final polymers show that intermolecular CTP was significant in all the polymerizations, which is typical for acrylate emulsion polymerizations performed in the absence of chain transfer agents, such as mercaptans [31] (which reduce crosslinking by providing a highly competitive H-abstraction chain termination mechanism).
The reason for lower instantaneous conversions in the sBA emulsion polymerization is not clear, though the type and level of inhibitor in this monomer, as supplied, was not specified (see Table S1), so it is possible that inhibitor removal was not effective for sBA. The lower instantaneous conversions in the emulsion polymerizations of nDA and iDA are most likely due to monomer transport limitations as a consequence of their very low water solubilities and, for nDA, artificial reduction of the conversion values arising from not taking into account loss of polymer in the form of coagulum, given the large amount of coagulum produced in the polymerization (see Table 2).
Effect of side-group length on CTP in emulsion polymerization of linear alkyl acrylates
A series of n-acrylates were studied with 1, 2, 4, 6, 8 and 10 carbons in the ester OR side-group in order to determine whether or not there is an effect of side-group length on the extent of CTP. With the exception of nDA, the conversion-time curves for the emulsion polymerizations were very similar, with only nOA showing slightly lower instantaneous conversions during the monomer feed (ca. 80% cf. 85–95% for the other acrylates; see Fig. S1). As described in the previous section, however, the nDA emulsion polymerization proceeded at much lower instantaneous conversions, the consequences of which are discussed when considering the %br data for these polymers.
The 13C NMR spectra of the final polymers have features that are identical to those for PnBA [7], except for the number of peaks in the δC 20–34 region, which arise from the different lengths of the aliphatic side-group, but do not interfere with the integrations of the branch Cq, CH and CH2 peaks. This is illustrated by the representative 13C NMR spectra shown in Fig. 3, which include PnBA as a reference point for previous work. The normal backbone CH2 and penultimate chain-end CH2 resonances show significant splitting due to the effects of tacticity, though this has no effect for the calculation of %br. Thus, for each of the polymers, %br could be calculated accurately from both the branch Cq and branch CH + CH2 peaks using Eqs. (2) and (3). The data are presented in Fig. 4, from which there is indication of a trend for a small increase in %br as the number of carbon atoms in the ester OR group (NsgC) increases, at least for NsgC in the range 1–6 (for which %br values increase from about 3.4 to about 4.7 mol%). For NsgC in the range 4–10, the trend is less certain because of the experimental error in measurement of %br. Nevertheless, recognizing that the instantaneous conversions during nDA emulsion homopolymerization were far lower than for the other n-acrylates, from inspection of Eq. (1), it is likely that the %br for PnDA is significantly below what it would have been if the reaction had proceeded with instantaneous conversions similar to those in the other polymerizations. In view of this, there is evidence for a continuous small increase in %br with NsgC by a factor of about 1.5 × for NsgC over the range 1–8.

Representative 13C NMR spectra of the final polymers from the series of n-alkyl acrylate emulsion homopolymerizations. The abbreviations used are: nb normal backbone; br branch; e chain-end; pe penultimate chain-end; sg side-group

Variation of %br with NsgC for the final polymers from the series of n-alkyl acrylate emulsion homopolymerizations: empty circle, %br from br Cq referenced to half the total backbone carbons, calculated using Eq. (2); and filled circle, %br from br CH + CH2 referenced to 3 × the total backbone carbons, calculated using Eq. (3)
The only previous study of branching for a series of n-alkyl acrylates investigated MA, EA, nBA and nHA solution polymerizations in toluene at 60 °C and employed quantitative 13C NMR to determine %br from the branch Cq peaks referenced only to the normal backbone CH peak (which will lead to small errors in %br that most probably lie within the experimental error for the measurements) [32]. Unlike in the present work, the %br values for the polymers were relatively low (1.3–2.3 mol%), most likely as a consequence of the lower reaction temperature and the use of solution polymerization, and showed no trend with NsgC, leading the authors to conclude that the OR side-group has no influence on %br (initial monomer concentration and conversion data were not provided, so their effects on the %br data cannot be assessed). In contrast, the higher extents of CTP in the monomer-starved emulsion homopolymerizations reported herein have facilitated the elucidation of a small increase of %br with NsgC. The trend does, however, comprise two influences based on Eq. (1), namely the rate coefficient ratios and the concentration terms, which need to be separated as far as is possible.
In order to access the molecular effects embodied in the rate coefficient ratios of Eq. (1), the effects arising from the concentration terms need to be removed and are accessible through the instantaneous conversions, which are mass based and so need to be converted to molar concentrations. For the intermolecular CTP term, the mass ratio [P]/[M] is equal to its molar ratio because the molar masses of the repeat unit and monomer are the same, so there is no adjustment needed to the trend shown in Fig. 4 if it is the dominant CTP process. However, even though [P]/[M] is high because the instantaneous conversions are high (explaining the high gel contents in the final polymers), in terms of kinetics, intramolecular CTP is expected to be dominant. Hence, for the purposes of the following discussion, we will consider only the intramolecular CTP term in Eq. (1), which contains the factor of 1/[M]. The value of [M] is controlled by the instantaneous conversion and by partitioning of monomer between the particle and aqueous phases; conversion of [M] from a mass to a molar concentration requires division by the monomer molar mass (Macrylate). Given that the instantaneous conversions in the emulsion homopolymerizations are similar for the majority of the acrylates, there will be a big effect of Macrylate in reducing the molar [M] as NsgC increases, though this will be compensated somewhat by an increase in partitioning of monomer into the particles as NsgC increases, an effect that will diminish quickly and should be significant only for NsgC ≤ 4. Accounting for partitioning is not feasible due to lack of detailed information on this, so its effects will need to be assumed as negligible. Although it is not possible to calculate accurate values of molar [M] because the many parameters required are not available, the underlying effects of NsgC on \({k}_{\mathrm{trP}}^{\mathrm{intra}}/{k}_{\mathrm{p}}\) can be revealed in terms of trends by normalising the %br data to the Macrylate values — we have chosen to use the molar mass of MA (MMA) as a reference value for Macrylate and to use MMA/Macrylate as the normalising factor for the %br data, i.e. by taking %br.(MMA/Macrylate), the effects of Macrylate on the trends in %br are eliminated (though in a very approximate way that also does not account for effects of overall conversion, for which Eq. (1) is not appropriate due to the assumptions in its derivation [22]). Figure 5 presents the normalised data, which show an opposite trend to that in Fig. 4, i.e. the molecular effect of increasing NsgC is to reduce \({k}_{\mathrm{trP}}^{\mathrm{intra}}/{k}_{\mathrm{p}}\).

Plot of %br data normalised by MMA/Macrylate to eliminate the effects of Macrylate on %br assuming that intramolecular CTP is dominant. The data are taken from Fig. 4 for the final polymers from the series of n-alkyl acrylate emulsion homopolymerizations: empty circle, %br from br Cq referenced to half the total backbone carbons, calculated using Eq. (2); and filled circle, %br from br CH + CH2 referenced to 3 × the total backbone carbons, calculated using Eq. (3)
In order to access the underlying trends in \(k_{\mathrm{trP}}^{\mathrm{intra}}\), values of kp are needed. PLP measurements have shown that the values of kp for n-alkyl acrylates and methacrylates increase (by about 3–4% per CH2) as NsgC increases [9, 33,34,35], though a definitive explanation for this observation is yet to be established. Most recently, taking into account all observations, the effect has been explained tentatively in terms of a reduced concentration of the ester CO2 group (that stabilises the chain radical in the transition state for propagation) as NsgC increases, thereby increasing the reactivity of the chain radical towards monomer and causing a consequent increase in kp [35]. Given that this interpretation of the effect is based on a thorough and well-considered analysis of the published data, we will assume it is correct for the purpose of discussing effects of the ester OR side-groups on CTP. The relative reduction in %br.(MMA/Macrylate) with NsgC is similar to the relative reduction expected in 1/kp as NsgC increases, which implies that \(k_{\mathrm{trP}}^{\mathrm{intra}}\) does not vary significantly with NsgC. Electronic effects on CTP are unlikely because the OR side-group is too remote from the sites of reaction to expect a strong electronic influence on reactivity. However, we did anticipate observing effects from steric influences on the probability of a chain adopting the 6-membered ring chain-end conformation that is necessary for intramolecular CTP to proceed (see Scheme 1b) because as NsgC increases, the volume swept out by rotations of the C–C bonds in the side-group creates greater free volume locally within the polymer coil that results in easier rotation of the backbone C–C bonds, as evidenced, for example, by a reduction in the glass transition temperature of the bulk polymer [36]. This greater local free volume within the polymer coil and the spatial requirement to accommodate the increasing size of the ester side-group as NsgC increases were expected to have an effect on the frequency with which the coil adopts the 6-membered ring chain-end conformation and the time for which it exists, once adopted, both of which could influence the frequency factor in the Arrhenius equation for \({k}_{\mathrm{trP}}^{\mathrm{intra}}\). Hence, it could be that the effects of NsgC on these two factors are self-compensating in terms of \({k}_{\mathrm{trP}}^{\mathrm{intra}}\), though the substantial uncertainties in the above analysis of the %br data make the hypotheses only tentative at best. Furthermore, as is evident from the high gel contents in the final polymers, intermolecular CTP is significant for these acrylate emulsion homopolymerizations, though the extent to which it contributes to %br is not known. Hence, the effect of NsgC on intermolecular CTP process should be considered briefly, for which the influences of NsgC on local free volume and backbone C–C bond rotation would be similar to those for intramolecular CTP; the major difference is that intermolecular CTP involves two polymer chains, so steric effects should be bigger, and the greater free volume in the coil as NsgC increases would be expected to reduce the overall steric barrier to an incoming chain radical.
Effect of side-group structure on CTP in emulsion polymerization of isomeric butyl acrylates
The isomeric butyl acrylates (NsgC = 4) were investigated in order to establish whether or not there is an effect of side-group isomerism on the extent of CTP for a relatively small side-group. With the exception of sBA, the emulsion homopolymerizations proceeded similarly with high instantaneous conversions (mostly > 90%) throughout (see Figs. 2 and S1). The much lower instantaneous conversions for sBA (as low as 52% soon after beginning the monomer feed, but mostly > 60%) resulted in a much lower gel content in the final polymer (36% compared to 70–77% for the other butyl acrylates), a strong indication that the extent of CTP had been reduced significantly by the lower instantaneous conversions, as would be expected from Eq. (1).
The 13C NMR spectra of the final polymers (see Figs. 3 and 6) have all the usual features with branch Cq and branch CH + CH2 peaks that are well resolved and facilitate accurate calculation of %br using Eqs. (2) and (3). The data are presented in Fig. 7 (since Macrylate is constant, there is no need to normalise the data). With the exception of PsBA, which has a significantly lower value of %br (3.2 mol%), PnBA, PiBA and PtBA have %br values (4.3–4.4 mol%) that are identical within experimental error. Given that the lower value of %br for PsBA almost certainly is a consequence of the lower instantaneous conversions during the polymerization, the results show that there is no measurable effect of side-group isomerism on %br for butyl side-groups. This is consistent with there being no effect of side-group isomerism on the values of kp determined by PLP for nBA [37] and tBA [38, 39], which have identical kp values at 80 °C, the temperature of polymerization used in our work. Clearly, butyl side-groups are too small for the changes in bulkiness between the isomers to have any major impact on the values of \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) and kp.

13C NMR spectra of the final polymers from iBA, sBA and tBA emulsion homopolymerizations. The abbreviations used are as given in Fig. 3

Variation of %br with side-group structure for the final polymers from isomeric butyl acrylate emulsion homopolymerizations: empty circle, %br from br Cq referenced to half the total backbone carbons, calculated using Eq. (2); and filled circle, %br from br CH + CH2 referenced to 3 × the total backbone carbons, calculated using Eq. (3)
Effect of side-group structure on CTP in emulsion polymerization of isomeric higher acrylates
In contrast to the observations for the butyl acrylates, the effect of side-group isomerism on %br for the higher acrylates (NsgC ≥ 6) was found to be significant. The emulsion polymerizations proceeded with similarly high instantaneous conversions, except for nDA and iDA (see Figs. 2 and S1), the reasons for which were discussed earlier. However, inspection of Table 2 reveals that the final polymer gel contents for nDA (73%) and iDA (67%) were in the same range as those for the other higher acrylate monomers (67–72%), indicating that the lower instantaneous conversions had much less of an effect on CTP than for sBA. This is consistent with the instantaneous conversions being much closer to those for the other acrylates for iDA compared to sBA and with the nDA conversions being higher than shown (see Fig. S1) because they are artificially reduced due to loss of polymer as coagulum.
Representative 13C NMR spectra of the final polymers are shown in Figs. 3 and 8. The branch Cq peak is observed in all the polymers, as are the branch CH + CH2 peaks with the notable exceptions of P2EHA, PnDA and PiDA. For P2EHA, the branch CH2 is obscured by a side-group peak, but the branch CH can still be resolved and so was used to calculate %br from Eq. (4). The 13C NMR spectra of PiOA and PiDA are complex because the monomers are mixtures of different non-linear isomeric esters, a consequence of which is that all backbone and branch CH and CH2 carbon peaks are hidden by side-group peaks; however, the branch Cq peak, though close to a side-group peak, is sufficiently well resolved to facilitate its use in determining %br by referencing its integral to that of the side-group OCHx peaks at δC 60–70. Hence, it was possible to determine %br accurately from the 13C NMR spectra for the all the acrylate homopolymers by one or more methods of calculation. The results from these calculations are presented in Fig. 9, which shows clearly that the polymers from the acrylates with non-linear side-groups have much higher %br (around 7 mol%) than those from the corresponding higher n-alkyl acrylate homopolymers (about 4.8 mol%), the only exception being P2EBA for which %br (about 4.7 mol%) is similar to the higher n-alkyl acrylate homopolymers. The values of %br for P2EHA, PcHA, PiOA and PiDA are the same within experimental error, despite the differences in side-group structure. Thus, simply having a large bulky OR side-group is sufficient to give a similar, relatively large, effect of increasing %br.

Representative 13C NMR spectra of the final polymers from emulsion homopolymerizations of acrylates with NsgC ≥ 6 and bulky OR ester side-groups. Spectra for both PiOA and PiDA are included because they show a high degree of complexity as a consequence of being mixtures of isomers. The abbreviations used are as given in Fig. 3

Variation of %br with side-group OR structure for the final polymers from emulsion homopolymerizations of the higher acrylates with NsgC ≥ 6: empty circle, %br from br Cq referenced to half the total backbone carbons, calculated using Eq. (2); empty square, %br from br Cq referenced to the total O–CHx side-group carbons, calculated using Eq. (5); and filled circle, %br from (i) br CH + CH2 referenced to 3 × the total backbone carbons, calculated using Eq. (3), for PnHA, PnOA, PnDA, P2EBA and PcHA and (ii) from br CH referenced to 1.5 × the total backbone carbons, calculated using Eq. (4), for P2EHA
Since Fig. 9 contains data for acrylates with different values of NsgC, it is appropriate to assume again that intramolecular CTP is dominant and to normalise the %br data by taking %br.(MMA/Macrylate) in order to eliminate approximately the effects of Macrylate and elucidate molecular effects on \({k}_{\mathrm{trP}}^{\mathrm{intra}}/{k}_{\mathrm{p}}\). The normalised %br data are shown in Fig. 10 (since MA is again used as the reference monomer, the ordinate scales of Figs. 5 and 9 are comparable). Despite the normalisation, for a given value of NsgC, the effect of changing from a linear to a non-linear ester OR group remains the same as in Fig. 9. Thus, there is a distinct effect of side-group non-linearity increasing \({k}_{\mathrm{trP}}^{\mathrm{intra}}/{k}_{\mathrm{p}}\). Unlike for the n-alkyl acrylates and methacrylates, measurements of kp by PLP for acrylates and methacrylates with non-linear OR side-groups have not shown definitive trends [34, 35]. There are, however, indications [35] for a slight increase in kp as NsgC increases for non-linear acrylates with NsgC > 8 and for a reduction in kp as the non-linearity of the OR side-group increases for acrylate isomers with NsgC = 13, the latter being explained tentatively in terms of steric influences of the greater non-linearity, i.e. more bulky side-groups, on rotational barriers. Arrhenius frequency factors and activation energies for propagation, determined by PLP, have been reported for some of the higher acrylates studied in the present work and give the values of kp at 80 °C summarised in Table 3, which also include nBA, iso-nonyl acrylate (iNA) and dodecyl acrylate (nDDA) as reference points.

Plot of %br data normalised by MMA/Macrylate to eliminate the effects of Macrylate on %br assuming that intramolecular CTP is dominant. The data are taken from Fig. 9 for the final polymers from emulsion homopolymerizations of the higher acrylates with NsgC ≥ 6: empty circle, %br from br Cq referenced to half the total backbone carbons, calculated using Eq. (2); empty square, %br from br Cq referenced to the total O–CHx side-group carbons, calculated using Eq. (5); and filled circle, %br from (i) br CH + CH2 referenced to 3 × the total backbone carbons, calculated using Eq. (3), for PnHA, PnOA, PnDA, P2EBA and PcHA and (ii) from br CH referenced to 1.5 × the total backbone carbons, calculated using Eq. (4), for P2EHA
Since kp for nHA and cHA are essentially the same at 80 °C, the large increase in %br.(MMA/Macrylate) from nHA to cHA shows that \(k_{\mathrm{trP}}^{\mathrm{intra}}\) must be much higher for cHA than for nHA (nominally by a factor of about 1.5). Comparison of the %br.(MMA/Macrylate) and kp data for cHA and 2EHA indicates that \(k_{\mathrm{trP}}^{\mathrm{intra}}\) for cHA also must be higher than for 2EHA homopolymerization (nominally by a factor of 1.3). Since values of kp for iOA and iDA are not known, the values of kp for iNA and nDDA are taken here as indicators of the magnitude of kp, which (from Table 3) are similar to the values of kp for nHA and cHA, but much higher than kp for 2EHA. Hence, to facilitate discussion, it will be assumed that kp values for iOA and iDA polymerization are similar to that for cHA. The values of %br.(MMA/Macrylate) for PiOA and PiDA are slightly lower than that for PcHA, so the effects of the mixed isomer esters in iOA and iDA are leading to an effect on CTP that is slightly less than for the cyclohexyl ester. Nevertheless, it is apparent that large bulky OR side-groups have a big effect of increasing \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) relative to that for the corresponding linear OR side-group. It seems unlikely that the origin of the effects of side-groups on \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) would be different for the series of acrylates with linear and non-linear OR side-groups studied here because they are all saturated hydrocarbon groups; hence, a common explanation of the observations needs to be sought. For the reasons described earlier, the observations will be considered again in terms of steric effects of the OR side-group. Compared to its linear equivalent, a non-linear OR side-group will be sterically bulkier and so is likely to impose greater restrictions on rotation of backbone C–C bonds, as well as sweeping out a smaller volume through C–C bond rotations within the side-group, thereby reducing the free volume within the polymer coil. The barrier to rotation of backbone C–C bonds with the non-linear OR side-groups will, therefore, be much greater, and the chain backbone will be considerably stiffer than for the equivalent linear OR side-group, so the frequency of adoption of the 6-membered ring chain-end conformation will be reduced, but its time of existence, once adopted, will increase. If our earlier hypothesis that these influences affect the frequency factor in the Arrhenius equation for \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) is correct, then the observations suggest that the ‘time of existence’ factor is more important for NsgC ≥ 6. The effect of side-group non-linearity on intermolecular CTP could well be different because it involves reaction between two polymeric species, for which the reduction in free volume in changing from a linear to a non-linear OR side-group may have a much greater effect.
Effect of temperature on chain transfer to polymer in methyl acrylate solution homopolymerization
As an addition to the studies of side-group effects, we also investigated the effect of temperature on the extent of CTP. It is well established that autoacceleration begins at low conversions in acrylate radical polymerizations and so removal of the heat of polymerization is necessary in order to keep local reaction temperatures controlled to the target reaction temperature. Thus, emulsion polymerization, which proceeds at high rates and is focused within latex particles, is not appropriate for studying the effect of temperature. Hence, solution polymerization was selected instead, with MA as the monomer because PMA has the simplest 13C NMR spectrum, which was expected to be an advantage for accuracy when analysing polymers with very low levels of branching. Methyl 2,2-dimethylpropionate was selected as the solvent because it does not undergo chain transfer to any significant extent. Since temperatures in the range 0–80 °C were to be studied, a redox couple of N,N-dimethyl-p-toluidine with benzoyl peroxide was used for initiation with the N,N-dimethyl-p-toluidine completely in the reaction vessel at t = 0 and the benzoyl peroxide fed as a solution in methyl 2,2-dimethylpropionate over a period of 60 min. The initial [MA] was 2.68 mol kg−1, and the polymerizations were taken to relatively high conversions in order to produce sufficient polymer for the analyses.
Representative 13C NMR spectra of the PMA samples produced are shown in Fig. 11. It is evident that determination of %br becomes subject to greater errors as its value decreases and that accurate removal of the contribution to the branch CH + CH2 integrals from the solvent Cq peak became more difficult as the level of branching reduced. By recording the 13C NMR spectra several times, the errors were estimated to be approximately: (i) ± 20% of the %br value for %br < 0.5 mol%; ± 15% of the %br value for 0.5 ≤ %br < 1.0 mol%; and ± 10% of the %br value for %br ≥ 1.0 mol%.

Representative 13C NMR spectra of the final polymers from solution homopolymerizations of MA in methyl 2,2-dimethylpropionate. The abbreviations used are as given in Fig. 3 plus: solv solvent
The results from the reactions are presented in Table 4. The value of Mn reduces as reaction temperature increases, as expected due to the combined effects of increasing temperature: (i) reducing the kinetic chain length in the absence of chain transfer (of any kind) [23, 24] and (ii) increasing CTP (i.e., ktrP/kp), which for intramolecular CTP (expected to be completely dominant under these solution polymerization conditions) will lead to reductions in Mn because the tertiary chain radicals resulting from CTP have substantially lower values of kp than for the normal secondary chain radicals [12, 13]. The values of Mw/Mn (2.5–3.2) show no trend and are much higher than the theoretical value of 1.5 for termination by combination [23, 24], which is the dominant mode of termination for acrylate radical polymerizations [25]; this is, at least in part, due to the high conversions and is common for acrylate homopolymers.
Thus, %br reduces as the reaction temperature reduces, as expected, and is not measurable for the PMA produced at 0 °C. Since all the 13C NMR spectra were recorded under fast pulse conditions, the %br values calculated from the branch Cq are much lower than those calculated from the branch CH + CH2 peaks. Although the results are not ideal for Arrhenius analysis because of the composite of rate coefficients that determine %br and the high conversions, Fig. 12 shows an Arrhenius plot of the %br data calculated from the branch CH + CH2 peaks (which, for the reasons discussed earlier, can be taken as quantitatively accurate). Analysis of the intercept in this plot cannot be justified, but the slope should approximate to (EtrP − Ep)/R where EtrP and Ep are the overall activation energies for CTP and propagation, respectively, and R is the gas constant. On this basis, taking the accepted value of Ep = 17.3 kJ mol−1 for MA [42], the slope of the plot gives EtrP = 40.7 kJ mol−1, which can be taken as the value for intramolecular CTP because intermolecular CTP is expected to make only a very minor contribution in these solution polymerizations of MA. Although a value of EtrP for intramolecular CTP in MA radical polymerizations has not been reported, the value obtained in the present work may be compared to those obtained for nBA radical polymerizations, for which a wide range of published values have been determined through experimental studies (29.3–47.7 kJ mol−1) [43]. Thus, the value of EtrP for MA obtained in this work is within the range reported for nBA.

Arrhenius plot of %br–T data for PMA from solution homopolymerization (where T is the absolute reaction temperature): empty circle, %br from br CH + CH2 referenced to 3 × the total backbone carbons, calculated using Eq. (3)
Summary and conclusions
Investigation of effects of 13C NMR conditions on %br values confirmed that, for most acrylate homopolymers, use of the branch CH and CH2 peak integrals referenced to the total backbone carbon integral from fast pulse spectra gives %br values (via Eqs. (3) and (4)) that are reliable and accurate with the benefit of requiring much less instrument time than for measurements done with NOE suppression and long pulse intervals (the exceptions in this work being PiOA and PiDA because their branch CH and CH2 peaks are masked by side-group peaks).
The study of MA solution homopolymerization at temperatures in the range 0–80 °C showed that Mn reduces as reaction temperature increases and that the broad MMDs, with Mw/Mn of 2.5–3.2, show no trend with temperature, all in accord with expectation. The extent of branching reduces as temperature reduces (%br is 2.1 mol% at 80 °C and reduces to a negligible level at 0 °C), approximate Arrhenius analysis of which gives an activation energy of 40.7 kJ mol−1 for intramolecular CTP (assumed to be dominant in solution polymerization). Although an approximation, this value falls within the wide range of experimentally measured values (29.3–47.7 kJ mol−1) reported for nBA.
Our studies of ester side-group effects on CTP have led to the following conclusions for effects of OR side-group structure on %br in the final polymers from monomer-starved semi-batch emulsion homopolymerizations of alkyl acrylates with NsgC in the range 1–10. The results have been analysed tentatively in terms of intramolecular CTP because this is still expected to be numerically the dominant CTP process, even though the high instantaneous conversions and high gel contents in the final polymers show that intermolecular CTP is significant.
For linear OR side-groups there is a small, continuous increase in %br as NsgC increases (from about 3.4 to 5.0 mol%). This arises predominantly from effects of Macrylate, which reduces molar [M] (because the polymerizations proceeded at similarly high instantaneous mass conversions) and the known (though small) increase in kp as NsgC increases. Although effects of NsgC on \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) were anticipated, no discernible effect is evident in the results.
For butyl acrylate isomers, there is no measurable effect of isomer structure on %br (4.3–4.4 mol%), indicating that butyl OR side-groups are too small for the changes in bulkiness between the isomers to have any major impact on the values of \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) and kp.
For OR side-groups with NsgC = 6, the effect of non-linearity is mixed, with nHA and 2EBA giving rise to polymers with similar %br (4.7–4.8 mol%), but cHA giving rise to polymer with a much higher %br (about 7.2 mol%). Thus, for nHA and 2EBA, as for the butyl acrylate isomers, there appears to be insufficient difference in bulkiness of the OR side-groups to have an effect on %br. However, the cyclohexyl side-group in cHA has a big effect of increasing %br, principally through steric effects increasing \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) because kp values for nHA and cHA are the same.
For OR side-groups with NsgC of 8 and10, there is a strong effect of side-group non-linearity increasing %br, with polymers from the linear acrylates having much lower %br (about 4.5–5.0 mol%) than those from the equivalent non-linear acrylates (about 6.7–7.1 mol%). Normalising the %br data for Macrylate effects on [M] does not change this trend. Since the kp values are expected to be similar for this group of acrylates, it is evident that the much bulkier non-linear OR groups cause a significant increase in \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) compared to the linear equivalents.
The observations have been considered in terms of steric effects of the OR side-group only because the side-group is too remote from the sites of reaction to expect a strong electronic influence on reactivity. We anticipated observing effects from established steric influences on local free volume in the polymer and on rotation of backbone C–C bonds, since they should affect the frequency with which the coil adopts the 6-membered ring chain-end conformation required for intramolecular CTP and the time for which that conformation exists, once adopted, both of which could influence the frequency factor in the Arrhenius equation for \({k}_{\mathrm{trP}}^{\mathrm{intra}}\). The only clear observation for an effect of side-group structure on \({k}_{\mathrm{trP}}^{\mathrm{intra}}\) is the increase arising from bulky non-linear OR side-groups compared to their linear equivalents for acrylates with NsgC ≥ 6. The bulkier non-linear OR side-groups will impose greater restrictions on rotation of backbone C–C bonds and a reduction of the free volume within the polymer coil, because C–C bond rotations within the side-group will sweep out a smaller volume. In terms of these hypotheses, the results suggest that increasing the time of existence of the 6-membered ring chain-end conformation may be the main effect of increasing the bulkiness of the side-group. However, these hypotheses are, at best, tentative and subject to substantial uncertainty. More detailed studies, both experimental and theoretical, will be needed in order to elucidate their origins with certainty. In order to better access the molecular effects as NsgC increases, dilute solution homopolymerizations performed at fixed initial molar [M] and taken to relatively low conversions are needed, but would have to be carried out on a large scale in order to generate sufficient polymer for analysis — intramolecular CTP is completely dominant in such reactions and gives relatively high %br (for any given acrylate) because the 1/[M] term in Eq. (1) is large [7, 17]. Since [M] would be known with accuracy and the intermolecular CTP term in Eq. (1) could be neglected, such experiments would provide a much better scrutiny of effects of NsgC on \({k}_{\mathrm{trP}}^{\mathrm{intra}}\), especially for the series of n-alkyl acrylates.
Despite the uncertainties around molecular interpretation of the results, the observations for the effects of side-group size and structure on %br in acrylate polymers produced by monomer-starved semi-batch emulsion polymerization are, without any doubt, real and significant. They are highly relevant for water-borne pressure-sensitive adhesives (copolymers produced using acrylates as the principal monomer) and offer insights into how adhesive performance could be modified through careful choice of acrylate monomers and polymerization conditions.
References
Scott GE, Senogles E (1970) Polymerization kinetics of n-lauryl acrylate. J Macromol Sci Chem A4:1105–1117
Scott GE, Senogles E (1973) Kinetic relationships in radical polymerization. J Macromol Sci Rev C9:49–69
Scott GE, Senogles E (1974) Polymerization kinetics of n-alkyl acrylates. J Macromol Sci Chem A8:753–773
Lovell PA, Shah TH, Heatley F (1991) Chain transfer to polymer in emulsion polymerisation of n-butyl acrylate studied by 13C NMR and GPC. Polym Commun 32:98–103
Lovell PA, Shah TH, Heatley F (1991) Chain transfer to polymer in emulsion polymerisation of n-butyl acrylate. Polym Mat Sci Eng 64:278–279
Lovell PA, Shah TH, Heatley F (1992) Correlation of the extent of chain transfer to polymer with reaction conditions for emulsion polymerization of n-butyl acrylate. Polymer Latexes: Preparation, Characterization and Applications. ACS Symposium Series, 492, Eds Daniels ES, Sudol ED, El-Aasser MS American Chemical Society Washington, DC 188–202
Ahmad NM, Heatley F, Lovell PA (1998) Chain transfer to polymer in free-radical solution polymerization of n-butyl acrylate studied by nmr spectroscopy. Macromolecules 31:2822–2827
Plessis C, Arzamendi G, Leiza JR, Schoonbrood HAS, Charmot D, Asua JM (2000) A decrease in effective acrylate propagation rate constants caused by intramolecular chain transfer. Macromolecules 33:4–7
Beuermann S, Buback M (2002) Rate coefficients of free-radical polymerization deduced from pulsed laser experiments. Prog Polym Sci 27:191–254
Yamada B, Tanaka K, Willemse R, van Herk AM (2002) Pulsed laser polymerization of cyclohexyl acrylate involving fragmentation of mid-chain radical. Polymer 34:692–699
Nikitin AN, Castignolles P, Charleux B, Vairon J-P (2003) Determination of propagation rate coefficient of acrylates by pulsed-laser polymerization in the presence of intramolecular chain transfer to polymer. Macromol Rap Commun 24:778–782
van Herk AM (2001) Pulsed initiation polymerization applied to acrylate monomers: sources of experimental failure. Macromol Rap Commun 22:687–689
van Herk AM (2009) Historic account of the development in the understanding of the propagation kinetics of acrylate radical polymerizations. Macromol Rap Commun 30:1964–1968
Barner-Kowollik C, Günzler F, Junkers T (2008) Pushing the limit: pulsed laser polymerization of n-butyl acrylate at 500 Hz. Macromolecules 41:8971–8973
Junkers T, Barner-Kowollik C (2008) The role of mid-chain radicals in acrylate free radical polymerization: branching and scission. J Polym Sci Polym Chem 46:7585–7605
Reyes Y, Arzamendi G, Asua JM, Leiza JR (2011) Branching at high frequency pulsed laser polymerizations of acrylate monomers. Macromolecules 44:3674–3679
Heatley F, Lovell PA, Yamashita T (2001) Chain transfer to polymer in free-radical solution polymerization of 2-ethylhexyl acrylate studied by NMR spectroscopy. Macromolecules 34:7636–7641
Plessis C, Arzamendi G, Alberdi JM, Agnely M, Leiza JR, Asua JM (2001) Intramolecular chain transfer to polymer in the emulsion polymerization of 2-ethylhexyl acrylate. Macromolecules 24:6138–6143
Chiefari J, Jeffery J, Mayadunne RTA, Moad G, Rizzardo E, Thang SH (1999) Chain transfer to polymer: a convenient route to macromonomers. Macromolecules 32:7700–7702
Koo SPS, Junkers T, Barner-Kowollik C (2009) Quantitative product spectrum analysis of poly(butyl acrylate) via electrospray ionization mass spectrometry. Macromolecules 42:62–69
Lena J-P, Deschamps M, Sciortino NF, Masters SL, Squire MA, Russell GT (2018) Effects of chain transfer agent and temperature on branching and β-scission in radical polymerization of 2-ethylhexyl acrylate. Macromol Chem Phys 219:1700579(1–15).
Ahmad NM, Charleux B, Farcet C, Ferguson CJ, Gaynor SG, Hawkett BS, Heatley F, Klumperman B, Konkolewicz D, Lovell PA, Matyjaszewski K, Venkatesh R (2009) Chain transfer to polymer and branching in controlled radical polymerizations of n-butyl acrylate. Macromol Rap Commun 30:2002–2021
Young RJ, Lovell PA (2011) Introduction to polymers, 3rd edn., CRC Press London.
Odian G (2004) Principles of polymerization, 4th edn., John Wiley & Sons Hoboken.
Moad G, Soloman DH (2006) The chemistry of radical polymerization, 2nd edn., Elsevier Oxford.
Farcet C, Belleney J, Charleux B, Pirri R (2002) Structural characterization of nitroxide-terminated poly(n-butyl acrylate) prepared in bulk and miniemulsion polymerizations. Macromolecules 35:4912–4918
Brydson JA (1999): Plastics materials, Butterworth-Heinemann Oxford.
Satas D (1989) Handbook of Pressure Sensitive Adhesive Technology, 2nd edn. Van Nostrand Reinhold, New York
de Gennes PG (1996) Soft adhesives. Langmuir 12:4497–4500
Creton C (1997) Materials science of pressure-sensitive adhesives Processing of Polymers. Materials Science and Technology 18. Ed Meijer EH Wiley-VCH Weinheim 707-741
Jovanović R, Dubé MA (2004) Emulsion-based pressure-sensitive adhesives: a review. J Macromol Sci Rev 44:1–51
Castignolles P, Graf R, Parkinson M, Wilhelm M, Gaborieau M (2009) Detection and quantification of branching in polyacrylates by size-exclusion chromatography (SEC) and melt-state 13C NMR spectroscopy. Polymer 50:2373–2383
Willemse R, van Herk AM (2010) Determination of propagation rate coefficients of a family of acrylates with PLP-MALDI-ToF-MS. Macromol Chem Phys 211:539–545
Haehnel AP, Schneider-Baumann M, Hiltebrandt KU, Misske AM, Barner-Kowollik C (2013) Global trends for kp? Expanding the frontier of ester side chain topography in acrylates and methacrylates. Macromolecules 46:15–28
Haehnel AP, Schneider-Baumann M, Arens L, Misske AM, Fleischhaker F, Barner-Kowollik C (2014) Global trends for kp? The influence of ester side chain topography in alkyl (meth)acrylates − completing the data base. Macromolecules 47:3483–3496
Sperling LH (2001) Introduction to physical polymer science, 3rd edn., Wiley-Interscience New York.
Asua JM, Beuermann S, Buback M, Castignolles P, Charleux B, Gilbert RG, Hutchinson RA, Leiza JR, Nikitin AN, Vairon J-P, van Herk AM (2004) Critically evaluated rate coefficients for free-radical polymerization, 5. Propagation rate coefficient for butyl acrylate. Macromol Chem Phys 205:2151–2160
Dervaux B, Junkers T, Schneider-Baumann M, Du Prez FE, Barner-Kowollik C (2009) Propagation rate coefficients of isobornyl acrylate, tert-butyl acrylate and 1-ethoxyethyl acrylate: a high frequency PLP-SEC study. J Polym Sci Polym Chem 47:6641–6654
Wenn B, Junkers T (2016) Kilohertz pulsed-laser-polymerization: simultaneous determination of backbiting, secondary, and tertiary radical propagation rate coefficients for tert-butyl acrylate. Macromol Rap Commun 37:781–787
Couvreur L, Piteau G, Castignolles P, Tonge M, Coutin B, Charleux B, Vairon J-P (2001) Pulsed-laser radical polymerization and propagation kinetic parameters of some alkyl acrylates. Macromol Symp 174:197–207
Junkers T, Schneider-Baumann M, Koo SPS, Castignolles P, Barner-Kowollik C (2010) Determination of propagation rate coefficients for methyl and 2-ethylhexyl acrylate via high frequency PLP-SEC under consideration of the impact of chain branching. Macromolecules 43:10427–10434
Barner-Kowollik C, Beuermann S, Buback M, Castignolles P, Charleux B, Coote ML, Hutchinson RA, Junkers T, Lacik I, Russell DR, Stach M, van Herk AM (2014) Critically evaluated rate coefficients in radical polymerization – 7. Secondary-radical propagation rate coefficients for methyl acrylate in the bulk. Polym Chem 5:204–212
Hamzehlou S, Ballard N, Reyes Y, Aguirre A, Asua JM, Leiza JR (2016) Analyzing the discrepancies in the activation energies of the backbiting and β-scission reactions in the radical polymerization of n-butyl acrylate. Polym Chem 7:2069–2077
Funding
The Engineering and Physical Sciences Research Council of the United Kingdom provided funding this work through research grants GR/R47356 and GR/R47363.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gray, I., Heatley, F. & Lovell, P.A. Effect of side-group structure and temperature on chain transfer to polymer and branching in acrylate homopolymerizations. Colloid Polym Sci 300, 445–463 (2022). https://doi.org/10.1007/s00396-021-04935-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-021-04935-1