
Abstract
Tachycardia-induced atrial fibrosis is a hallmark of the structural remodeling of atrial fibrillation (AF). The mechanisms underlying tachycardia-induced atrial fibrosis remain unclear. In our previous study, we found that Smad7-downregulation promoted the development of atrial fibrosis in AF. Fibroblasts are enriched in microRNA-21 (miR-21), which contributes to the development of fibrosis and heart failure in the cardiovascular system. Our study was designed to test the hypothesis that miR-21 reinforces the TGF-β1/Smad signaling pathway in AF-induced atrial fibrosis by down-regulating Smad7. Rapid atrial pacing (RAP, 1000 ppm) was applied to the left atrium of the rabbit heart to induce atrial fibrillation and fibrosis. qRT-PCR and northern blot analysis revealed that RAP caused a marked increase in the expression of miR-21. Transfection with a miR-21 inhibitor significantly increased the expression of Smad7, while the expression of collagen I/III significantly decreased. These changes were implicated in the AF-induced release of miR-21 and down-regulation of Smad7. Adult rat cardiac fibroblasts treated with TGF-β1 showed increased miR-21 expression and decreased Smad7 expression. Pretreatment with a TGF-β1 inhibitor reduced the TGF-β1-induced up-regulation of miR-21. Pretreatment with pre-miR-21 and a miR-21 inhibitor significantly decreased and increased Smad7 expression, respectively. This result was negatively correlated with the expression of collagen I/III in fibroblasts. Moreover, the results of a luciferase activity assay suggest that Smad7 is a validated miR-21 target in CFs. Our results provide compelling evidence that the miR-21 specific degradation of Smad7 may decrease the inhibitory feedback regulation of TGF-β1/Smad signaling and serves as a new insight of the mechanism of atrial fibrosis in atrial fibrillation.
Similar content being viewed by others


Introduction
Atrial fibrillation (AF) is one of the most common cardiac arrhythmias and remains a major contributor to morbidity and mortality in clinical practice [1, 2]. Myocardial fibrosis plays a key role in the development and maintenance of AF [3, 4]. A better understanding of the mechanisms underlying myocardial fibrosis in AF may contribute to the more effective prevention and treatment of AF in the future.
Myocardial fibrosis has been proved to be the main cause of atrial fibrillation [3, 5]. Atrial fibrosis causes intra- and inter-atrial inhomogeneity in conduction, thus creating a substrate for local re-entry and contributing to the maintenance and progression of AF [6–8]. Our previous study clearly indicated that atrial fibrosis plays an important role in the induction and perpetuation of AF [9, 10]. The transforming growth factor-β1 (TGF-β1)/Smad signaling pathway is one of the most classic signaling pathways involved in the regulation of fibrosis [9, 10]. In the pathological conditions of AF, TGF-β1 synthesis and secretion is increased [3, 10, 11]. Activated TGF-β1 binds to membrane-bound heteromeric receptor kinases (TβRI and TβRII) that transduce intracellular signals via both Smad and non-Smad pathways. Activated TβRI and TβRII receptors phosphorylate receptor regulated Smads (R-Smads, such as Smad2/3), which form homomeric complexes and heteromeric complexes with a co-Smad (Smad4) [8, 9, 12]. These activated Smad complexes translocate into the nucleus, where they accumulate and bind to target genes to directly regulate their transcription and the production of related proteins such as fibrin [5, 8]. The inhibitory Smad proteins (I-Smads, such as Smad6/7) are capable of inhibiting TGF-β1 signaling. Smad7 binds to activated TβRI and prevents the phosphorylation of Smad2/3 or recruits the ubiquitin ligases Smurf1 and Smurf2 to induce proteasomal degradation of the receptor complexes. Therefore, Smad7 may act as a major negative regulator, forming autoinhibitory feedback loops in the TGF-β1/Smad2/3 signaling pathway [11, 12]. We previously reported that the high expression of TGF-β1 in pathological conditions could cause the down-regulation of Smad7, which contributes to the development of myocardial fibrosis in AF [10]. However, the molecular mechanism of the regulation of Smad7 expression through TGF-β1 remains unclear in AF.
MicroRNAs (miRNAs) are a broad class of small non-coding RNAs that control the expression of complementary target messenger RNAs. MicroRNA-21 (miR-21) is consistently reported to be upregulated in both cancer and various forms of cardiovascular disease [13, 14]. Fibroblasts in the cardiovascular system are enriched in miR-21, which contributes to the development of fibrosis and heart failure [15]. miR-21 has therefore emerged as an interesting candidate for the development of therapeutic strategies against many forms of heart disease. The inhibition of miR-21 by synthetic miRNA antagonists improved heart function in a cardiac disease model [16–19]. The same beneficial effects were observed in miR-21 knockout mice subjected to pressure-overload of the left ventricle, underlining the potential of miR-21 as a therapeutic target [20]. In recent years, additional roles of miR-21 in cardiovascular and pulmonary diseases have been described, including cardiac and pulmonary fibrosis as well as myocardial infarction [21–23]. It has been reported that TGF-β1 can increase miR-21 expression in cardiac fibroblasts and may be involved in the regulation of myocardial fibrosis [22, 24]. Animal experiments found that miR-21 expression affected the TGF-β1-induced renal tubular epithelial-to-mesenchymal transition in diabetic nephropathy by regulating Smad7 [25]. However, mechanistic data on the relationship between miR-21 and the TGF-β1/Smad7 signaling pathway in AF are still missing. In the present study, our aim was to determine if miR-21 up-regulation could enhance TGF-β1-induced myocardial fibrosis by inhibiting Smad7.
Materials and methods
Ethical standards
This study complied with the Declaration of Helsinki and the ethics committee of Guangdong Cardiovascular Institute, Guangdong General Hospital approved the study protocol. Informed consent from all participants was obtained before the initial coronary angiography. All of the experimental procedures were performed according to the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and were in compliance with the guidelines specified by the Chinese Heart Association policy on research animal use and the Public Health Service policy on the use of laboratory animals.
Animals
Male New Zealand rabbits (2.0–2.5 kg) were randomly divided into 4 groups (n = 10 for each group): normal control (CR), sham control (SH), rapid atrial pacing (RAP) and RAP + miR-21 inhibitor(the miR-21 inhibitor was administered to this group through a lentiviral vector, Genechem, Shanghai, China). All of the rabbits except for one in the RAP group survived to the end of the study. The data generated from the dead rabbit was not included in the results presented in this paper.
Animal model and experimental design
All the rabbits were intravenously anesthetized with 30 mg kg−1 pentobarbital sodium before being intubated and placed on mechanical ventilation with a volume-cycled ventilator (Model HX-200, TAIMENG, Chengdu, China). The heart was exposed at the center of the breastbone and a custom-designed set of electrodes, comprising a pair of electrodes with a distal hook for pacing and a pair of electrodes was sutured to the epicardial surface of the left atrium. The electrodes contain an interelectrode distance of 15 mm aligned proximally that serves for recording. The distal ends of these electrodes leads were tunneled subcutaneously and exposed at the animal’s back, where the electrodes were then connected to a pacemaker (output of 6 V with 1.0 ms pulse duration, Guangzhou Academy of Sciences, China) in the jacket. The pacemaker was programmed to provide RAP at 1000 ppm, and this pacing rate was maintained continuously for 4 weeks with a brief breaking period for the measurement of electrophysiological and mechanical parameters (Supplementary Table 1) [10, 26]. The rabbits in Group CR were not subjected to surgery while those in Group SH (sham control) experienced identical surgical procedure, but no RAP. When surgery was completed, the rabbits were given antibiotics and allowed to recover for 5 days. Postoperative care included the administration of antibiotics and analgesics.
MiR-control lentivirus vectors (109 TU/ml, 4 μl/day) and miR-21 inhibitor lentivirus vectors (109 TU/ml, 4 μl/day) were injected 2 times/per week for 4 weeks. The transcript for the miR-21 inhibitor was PCR purified and inserted into the lentiviral vector under the control of the CMV promoter. The GFP reporter gene was inserted at the 3′ end of the miR-21 inhibitor gene. Lentiviral vectors pseudotyped with the VSVg coat were produced. One day prior to pacing, the miRNA lentiviral vectors were injected into the jugular vein. At the end of the experiment, all of the rabbits were euthanized, and the left atria were examined histopathologically and collected for mRNA and protein analysis.
Masson trichrome staining for collagen
Masson trichrome staining of the paraffin section prepared from the Bouin-fixed samples was performed as previously described [27]. Adobe Photoshop 5.5 and Scion Images for Windows Beta 4.0.2 software were used for quantitate atrial collagen content [25].
Cell culture and transfection
The experimental procedure to isolate cardiac fibroblasts was approved by the Animal Care Committee of Guangzhou, China and was performed according to the method of Meszaros et al. Briefly, the ventricles of 2–3 hearts from adult male Sprague–Dawley rats (250–300 g) were minced, pooled, and placed in a collagenase/protease (Sigma, St Louis, MO, USA) digestion solution. The cells dissociated in the first treatment were discarded. After three digestions lasting 6 min each, the cells were pooled. The debris and myocytes were removed by unit gravity sedimentation. The fibroblasts were suspended in Dulbecco’s Modified Eagle Medium (DMEM) containing 1 % penicillin/streptomycin and 10 % fetal bovine serum. After a 60-min period of attachment to uncoated culture plates, those cells that were weakly attached or unattached were rinsed free and discarded. The attached cells were cultured in an incubator with 5 % CO2 at 37 °C. The culture medium was changed every other day. These cultures contained N95 % cardiac fibroblasts (CFs) as indicated by positive vimentin expression and negative myoactin expression. Those cells in passages 2 and 3 were used in the following studies.
Cells were plated on 60 mm2 plates at a density of 1 × 106 cells per plate. When the cells reached 70–80 % confluency, they were transfected with pre-miR-21 or control pre-miR (Ambion, Austin, TX, USA) using the TransIT TKO transfection reagent (Mirus Bio, Madison, WI, USA). To investigate the role of miR-21 in TGF-β1-induced collagen expression, we performed miR-21 transfection experiments in CFs. For these experiments, the cells were seeded at a density of 2 × 104 cells/cm2 in serum-free DMEM/F12. In this study, the cells were divided into the following groups: cells without transfection (blank control group, Group CR), cells transfected with the miR-control lentiviral vector (30 μM) (miR-control group, Group M), cells transfected with the pre-miR-21 (miR-21 over-expression) (30 μM) lentiviral vector (pre-miR-21 group, Group PM), cells treated with 10 ng/ml TGF-β1 (TGF-β1 group, Group T), cells treated with 10 ng/ml TGF-β1 plus the pre-miR-21 vector (30 μM) (TGF-β1 + pre-miR-21 group, Group TP), and cells treated with 10 ng/ml TGF-β1 plus the miR-21 inhibitor (30 μM) (TGF-β1 + miR-21 inhibitor group, Group TI).
Real-time PCR
Total RNA was extracted with TRIzol reagent (Gibco-BRL Life Technologies, New York, USA). cDNA was synthesized with the SYBR ExScript RT-PCR kit (TOYOBO, Japan) according to the protocol provided by the manufacturer. PCR primers for TGF-β1 (forward: 5′-ACA TTG ACTTCC GCA AGG AC-3′; reverse: 5′-TAG TAC ACG ATG GGC AGTGG-3′), Smad7 (forward: 5′-GTG GCA TAC TGG GAG GAGAA-3′; reverse: 5′-GAT GGA GAA ACC AGG GAA CA-3′), collagen I (forward: 5′-TGG CAA GAA CGG AGA TGAC-3′; reverse: 5′-TCC AAA CCA CTG AAA CCT CTG-3′), and collagen III (forward: 5′-TTC CTT TGT GGG CTG TGT CT-3′; reverse: 5′-TTG GCT TCT CTC ACT TTC CAG-3′) were designed with Primer Express software (Applied Biosystems). GAPDH (forward: 5′-GCA CCG TCA AGG CTG AGA AC-3; reverse: 5′-ATG GTG GTG AAG ACG CCA GT-3′) was used as a reference to normalize the input amount of RNA for all samples. Real-time PCR was performed using an ABI7300 Real-Time PCR system (Applied Biosystems) with the SYBR green fluorophore. All of the reactions were performed in at least duplicate for every sample. Threshold cycle (Ct) data were collected using the Sequence Detection Software version 1.2.3 (Applied Biosystems). The fold change in mRNA level relative to GAPDH was calculated using the 2∆∆Ct method [28].
TaqMan MicroRNA Reverse transcription reactions and TaqMan MicroRNA quantitative polymerase chain reactions (qPCR) were performed to detect miR-21 and an endogenous control, RNA U6 small nuclear (RNU6B) expression using the MicroRNA TaqMan Reverse Transcription Kit and the TaqMan MicroRNA Assays (Applied BioSystems, Carlsbad, CA, USA) according to manufacturer’s instructions. The miRNA detection conditions were: 95 °C for 10 min and 40 cycles of 95 °C for 15 s, 60 °C for 1 min. The miRNA expression levels were calculated as the cycle threshold (-delta CT) of miR-21 and normalized with an endogenous control. MiR-21 (forward: 5′-GCA CCG TCA AGG CTG AGA AC-3; reverse: 5′- CAG CCC ATC GAC TGG TG-3′) and RUN6 (forward: 5′-CTC GCT TCG GCA GCA CA-3; reverse: 5′- AAC GCT TCA CGA ATT TGC GT-3′) were used as a reference to normalize the input amount of RNA for all samples. Real-time PCR was performed using an ABI7300 Real-Time PCR system (Applied Biosystems) with the SYBR green fluorophore. All of the reactions were performed in at least duplicate for every sample. Threshold cycle (Ct) data were collected using the Sequence Detection Software version 1.2.3 (Applied Biosystems). The fold change in mRNA level relative to GAPDH was calculated using the 2∆∆Ct method [29].
Northern blotting
RNA was extracted by standard trizol method and suspended in DNAse- and RNAse-free water. Samples were run on 15 % 7 M urea-acrylamide gels. Electrophoresis was performed at 20 mA for about 3 h at room temperature. The oligonucleotides to be used as probes were purchased from Sigma (Milan, Italy). After electrophoresis on urea-acrylamide gels, the RNA was transferred to hybond nylon filters at 10 V overnight at 4 °C. After the ultraviolet cross-linking, the filters were prehybridized in 6 X SSPE, 5 X DENHARDT and 0.5 % SDS solution. Ten picomoles probes were radiolabelled using P32 γATP by standard reaction at 37 °C. Microspin G25 columns (Amersham) were used to separate radiolabelled probe from unincorporated γATP. The prehybridization and hybridization were performed in the same buffer, at 37 °C overnight. The filters were washed once in prewarmed washing solution containing 6 X SSPE and exposed in cassettes containing phosphor screen overnight. The signals were read by Amersham Typhoon 9200 phosphoimager and densitometry was performed with IMAGEQUANT software (Amersham). The filters were stripped in a solution containing 0.2 × SSPE and 0.2 % SDS for 10 min at 95 °C and subsequently rehybridized with another probe [30].
Luciferase reporter gene assays
To examine whether Smad7 is a valid target of miR-21, a single copy of the putative miR-21-recognition element from the 3′-UTR of the Smad7 gene was cloned into the GV306 plasmid vector downstream of the dual luciferase reporter gene (Genechem, Shanghai, China). CFs were co-transfected with the GV306 vector containing the Smad7 3′-UTR and the miR-21 overexpression plasmid using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Co-transfection with a non-targeting negative control RNA was performed as a control. At 48 h post transfection, the cells were lysed and assayed for luciferase activity with a dual-luciferase reporter assay kit (Promega, Madison, WI, USA) on a luminometer (Lumat LB9507).
Immunohistochemistry
After fixed in 10 % neutral formalin for 24 h, the middle ring of left atrium was embedded in paraffin and cut into 4 μm thick sections. The section was incubated with primary antibodies (goat polyclonal anti-Smad7 antibody from Santa Cruz Biotechnology at a 1:150 dilution) at 4 °C overnight. Incubation with the secondary antibody (biotinylated rabbit anti-goat antibody also from Boster Biotect CO). was performed at 37 °C for 30 min. Staining was generated with the streptavidin biotin-peroxidase complex immunohistochemical staining kit from Boster Biotect CO. Negative controls were performed by leaving out the primary antibody. Sections were viewed with 400× magnification and the intensity of the collagen staining was analyzed using the IBAS2.5 Image Analytical System (Institute of Biomedical Engineering, Beijing, China) as described before (Sheng, 1995). Five random fields per section were analyzed to produce a single number for the section [31].
Immunocytochemistry
The experimental procedure was performed as described previously [9], the cells were incubated with a primary antibody (anti-vimentin, myoactin, or Smad7) (1:100) in 5 % BSA overnight at 4 °C. The cells were then washed and incubated with a rhodamine-conjugated secondary antibody (1:500). The intensity of the Smad7 staining was analyzed using the IBAS2.5 Image Analytical System (Institute of Biomedical Engineering, Beijing, China) as previously described [10].
Western blot
Western blot was performed as described previously [12]. The primary antibodies (Santa Cruz Biotechnology) were diluted as follows: TGF-β1, 1:200; Smad7, 1:250; collagen I, 1:200; collagen III, 1:200; and β-actin, 1:500. Horseradish peroxidase–labeled secondary antibodies (Cell Signaling Technology) were diluted 1:1000 with 0.2 % TBS-T and 1 % skim milk. The protein bands on the Western blots were visualized using ECL Plus (Amersham, Arlington Heights, IL, USA). The relative band densities were normalized against β-actin.
Statistical analysis
The data are expressed as mean ± SE. ANOVA and the Student’s t test were used to determine statistical significance. A two-tailed probability value of 0.05 was considered statistically significant.
Results
MiRNA-21 expression profile in atrial tachypacing-induced rabbit model of AF
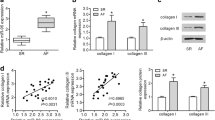
As shown in Fig. 1a, qRT-PCR analysis confirmed that miR-21 was elevated by 4.5 times in RAP samples over control samples (Fig. 1a). The expression of endogenous miR-21 in the heart and the up-regulation of miR-21 were verified with Northern blot analysis (Fig. 1b, c). These findings prompted us to focus on exploring the role of miR-21 in AF and the associated atrial remodeling. Real-time PCR and quantitative Western blot analysis found that the expression of TGF-β1 mRNA and protein (Fig. 1d–f) was significantly higher in the left atrium of group RAP than with the non-paced group CR and group SH. However, the expression of the inhibitory Smad, Smad7, was significantly lower in the RAP group (Fig. 1e, f). This finding further confirmed that TGF-β1/Smad7 was involved in RAP-induced myocardial remodeling. Furthermore, through the computational prediction of target genes, we identified Smad7 as a potential target for miR-21. These findings prompted us to focus on exploring the role of miR-21 in AF and the association with Smad7 expression.

MiRNA expression profiling in atrial samples from a rabbit model of AF. a Quantitative real-time RT-PCR (qRT-PCR) verification of the miRNA expression profile in pacing rabbits. b Representative Northern blot depicts the expression of miR-21 mRNA. c Mean miR-21 expression levels in the control group (CR), sham group (SH), and rapid atrial pacing group (RAP) groups. RAP caused a significant increase in miR-21 expression. d Quantitative analysis of TGF-β1 and Smad7 expression by real-time RT-PCR. The relative TGF-β1/Smad7 mRNA levels (TGF-β1/GAPDH or Smad7/GAPDH in arbitrary units) were normalized to the expression levels of group CR and group SH and the relative expression levels (in fold expression) were calculated. e Representative Western blot gel depicting the levels of TGF-β1 and Smad7 protein. f Mean TGF-β1 and Smad7 protein levels in the control group (CR), sham group (SH), and RAP group (RAP) groups. RAP caused a significant increase in expression of TGF-β1, but decreased the expression of Smad7 (*p < 0.05 vs. CR, unpaired t test; n ≥ 9 independent samples for each group)
Down-expression of miR-21 ameliorates myocardial fibrosis in vivo
On the basis of the above findings, we decided to examine whether reduced expression of miR-21 ameliorates myocardial fibrosis in AF. First, miR-21 inhibitor lentiviral vectors were injected into the jugular vein of rabbits 2 times per week for 4 weeks. RAP began one day after the injections and lasted for 4 weeks. At the end of the experiment, we found that miR-21 expression was significantly lower in the hearts of treated animals (Fig. 2a–c, p < 0.05), suggesting that miR-21 inhibitor lentiviral vectors can inhibit miR-21 expression in the heart. Next, we found that the incidence of atrial fibrillation after transfection with the miR-21 inhibitor lentiviral vector decreased from 89 % in the RAP group to 50 % in the lentiviral group (8 of 9 in the RAP group, 5 of 10 in the RAP+miR-21 inhibitor group). Furthermore, as shown in Table 1 and Fig. 2e, f, RAP increased the hydroxyproline content, collagen I and collagen III deposition, left atrial weight, and left atrial mass index of the animals’ hearts (Table 1). Transfection with the miR-21 inhibitor caused a significant decrease in hydroxyproline content, collagen I and collagen III deposition, left atrial weight, and left atrial mass index and attenuated the progression of RAP-induced atrial fibrosis. These data suggest that RAP-induced atrial fibrosis may be ameliorated by blocking miR-21 in the heart.

The effect of a miR-21 inhibitor in RAP-induced myocardial fibrosis in vivo. a miR-21 mRNA levels were examined by qRT-PCR. The miR-21 inhibitor decreased miR-21 expression in the heart of experimental rabbits (rabbits were treated with the miR-21 inhibitor and RAP for 4 weeks). b Representative Northern blot showing miR-21. c Mean miR-21 expression levels in the CR group and the RAP+miR-21 inhibitor group. d Mean incidence of AF. In contrast to the RAP group (89 % incidence of AF), treatment with the miR-21 inhibitor decreased the incidence of AF to 50 % (*p < 0.05). e Masson trichrome staining of rabbit atrial samples from the CR, RAP, and RAP+miR-21 inhibitor groups. The myocardium was stained red and collagens were stained blue. Bar 50 μm. f Mean collagen content in the left atria of the 3 groups (n ≥ 9 per group). RAP caused significant collagen deposition in the left atria that was attenuated by the miR-21 inhibitor (*p < 0.05 vs. CR, # p < 0.05 vs. RAP, unpaired t test; n ≥ 9 independent samples for each group)
Next, Smad7 and collagen I/III mRNA and protein were detected by qRT-PCR and Western blotting. Smad7 mRNA and protein levels were significantly lower than the CR group (Fig. 3a–c, p < 0.05). Conversely, collagen I/III mRNA and protein were significantly higher in the RAP group than the CR group (Fig. 3f–h, p < 0.05). Furthermore, immunohistochemical staining indicated that RAP induced the down-regulation of Smad7 (Fig. 3d, e, p < 0.05). More importantly, Smad7 expression increased after transfection with miR-21 inhibitor, whereas collagen I/III decreased (Fig. 3, p < 0.05). These data show that the miR-21 inhibitor could inhibit myocardial fibrosis by up-regulating Smad7 expression in vivo.

Effect of the miR-21 inhibitor on RAP-induced Smad7 and collagen I/III expression in vivo. a Quantitative analysis of Smad7 expression by qRT-PCR. The relative Smad7 mRNA expression ratio (Smad7/GAPDH in arbitrary units) in the left atria was normalized to the expression in group CR, and the relative expression levels (in fold expression) were calculated. b Representative Western blot depicts the expression of Smad7 protein. c Mean Smad7 protein levels in the CR, RAP and RAP + miR-21 inhibitor groups. The level of Smad7 was significantly lower in the left atria of the RAP group. After treatment with the miR-21 inhibitor, the level of Smad7 increased significantly compared with the CR group and the RAP group. d Representative immunohistochemical staining images for Smad7 (magnification: ×400). e Graphical representation of the abundance of Smad7 protein in the sections. f Quantitative analysis of the expression of collagen I and III transcripts by qRT-PCR. The relative collagen I and III mRNA levels (collagen I and III/GAPDH in arbitrary units) in the left atria were normalized to the levels in group CR and the relative expression levels (in fold expression) were calculated. g Representative Western blot depicts the expression of collagen I and III protein. h Mean collagen I and III protein levels in the CR, RAP and RAP + miR-21 inhibitor groups. The collagen I and III level was significantly higher in the left atria of the RAP group. After treatment with the miR-21 inhibitor, the level of collagen I and III decreased significantly compared with the CR and RAP group (*p < 0.05 vs. CR; # p < 0.05 vs. RAP, n ≥ 9 independent protein samples for each group)
Effect of TGF-β1 on miR-21 expression in vitro
Earlier reports have shown that miR-21 is extensively involved in myocardial fibrosis, and miR-21 is considered to be associated with TGF-β1-induced myocardial fibrosis [22, 32]. However, there are few reports about the role of miR-21 in AF. We have reported that TGF-β1 is up-regulated and promotes myocardial fibrosis in AF. In this study, we explored the relationship between miR-21 and TGF-β1 in cardiac fibroblasts.
First, miR-21 expression in the presence of TGF-β1 (10 ng/ml) was examined by qRT-PCR and Northern blot. We found that TGF-β1 can enhance miR-21 expression (Fig. 4a–c, p < 0.05). Next, we investigated whether miR-21 over-expression promoted the TGF-β1-induced expression of collagen I/III, which were detected by qRT-PCR and Western blot. Before treating the cells with TGF-β1, CFs were transfected with miR-control and miR-21 over-expression lentiviral vectors. miR-21 over-expression increased collagen I/III mRNA and protein levels. Meanwhile, CFs were treated with TGF-β1, with the results indicating that TGF-β1 could increase collagen I/III mRNA expression (Fig. 4h, i). Next, CFs were transfected with miR-control and miR-21 over-expression lentiviral vectors before being transfected with TGF-β1 (10 ng/ml) for 48 h. TGF-β1 plus miR-21 overexpression enhanced collagen I/III mRNA expression more significantly than TGF-β1 treatment alone (Fig. 4h, i). Interestingly, we found that miR-21 inhibition significantly weakened TGF-β1-induced collagen I/III mRNA expression in the CFs that were treated with TGF-β1 plus the miR-21 inhibitor. Furthermore, the effect on collagen I/III proteins was consistent with the effect on these mRNAs (Fig. 4j, k). Taken together, our results demonstrate that TGF-β1-induced collagen I/III expression might result from the expression of miR-21 in vitro.

Effect of miR-21 on TGF-β1-induced collagen I/III expression. a qRT-PCR demonstrating that TGF-β1 (10 ng/ml) and TGF-β1 inhibitor(10 μg/ml) induces and decrease miR-21 expression at 48 h. b Representative Northern blot showing miR-21. c Values of miR-21 expression are the mean ± SE. d Northern blot verifying the upregulation of miR-21 in rats fibroblast after transfection with pre-miR-21. e qRT-PCR demonstrating that miR-21 levels increased in rat fibroblasts after transfection with pre-miR-21. f Northern blot verifying the down-regulation of miR-21 in rat fibroblasts after transfection with the miR-21 inhibitor. g qRT-PCR demonstrating that miR-21 levels deceased in rat fibroblasts after transfection with the miR-21 inhibitor. h, i Quantitative analysis of collagen I and III expression by qRT-PCR. The relative collagen I and III mRNA level (collagen I and III/GAPDH in arbitrary units) in the left atria was normalized to the expression in group CR and the relative expression levels (in fold expression) were calculated. j Representative Western blot depicting he expression of collagen I and III protein. k Mean collagen I and III levels in the control (CR), miR-control (M), pre-miR-21 (PM), TGF-β1 (T), TGF-β1 + pre-miR-21 (TP) and TGF-β1 + miR-21 inhibitor (TI) groups (*p < 0.05 vs. CR, # p < 0.05 vs. T, unpaired t test; n = 10 independent samples for each group)
Over-expression of TGF-β1 induces Smad7 down-regulation through miRNA-21
Our results indicated that TGF-β1-induced collagen I/III expression might occur through the up-regulation of miR-21, meanwhile we found in our previous studies that the over-expression of TGF-β1 induced the degradation of Smad7 which may decrease the inhibitory feedback regulation of TGF-β1/Smad signaling pathway, so we sought to determine the relationship between miR-21 and Smad7 in TGF-β1-induced collagen expression. According to the TargetScan (http://www.targetscan.org/) and Pictar databases (http://pictar.mdcberlin.de/) and previous experiments, Smad7 is a validated target of miR-21 [25, 33]. Therefore, to further confirm that Smad7 is a validated miR-21 target in CFs, we performed luciferase reporter assays. The expression of the wild-type luciferase-Smad7-3′UTR reporter was remarkably lower than the mutant luciferase-Smad7-3′UTR reporter and control plasmid (Fig. 5a, b), suggesting that Smad7 is a validated miR-21 target in CFs and that miR-21 can inhibit Smad7 expression, leading to the further amplification of TGF-β1 signaling. Next, we investigated the effect of miR-21 over-expression on Smad7 mRNA and protein, which were detected by RT-PCR and western blot. Before treatment with TGF-β1, CFs were transfected with the miR-control and miR-21 over-expression lentiviral vectors. In this assay, we found that miR-21 over-expression decreased Smad7 mRNA levels. Next, we tested the effect of miR-21 overexpression on TGF-β1-induced Smad7 down-regulation. Prior to the transfection, we confirmed that TGF-β1 decreased Smad7 mRNA expression in CFs. Next, TGF-β1-treated CFs were transfected with the miR-control and miR-21 over-expression lentiviral vectors. The results of this assay demonstrated that miR-21 over-expression significantly enhanced TGF-β1-induced Smad7 down-regulation compared with the TGF-β1 group and the miR-control group (Fig. 5d). Interestingly, we found that miR-21 inhibition significantly weakened TGF-β1-induced Smad7 mRNA down-regulation in the CFs that treated with TGF-β1 plus the miR-21 inhibitor (Fig. 5d). Moreover, the effect on the Smad7 proteins was in line with the effect on these mRNAs (Fig. 5e, f). Immunocytochemistry showed that TGF-β1 decreased the nuclear and perinuclear accumulation of Smad7. The miR-21 inhibitor, but not pre-miR-21, increased the accumulation of Smad7 (Fig. 5c). Taken together, our data show that miR-21 over-expression directly down-regulates Smad7 expression.

Effect of miR-21 on the TGF-β1-mediated down-regulation of Smad7. a Alignment of hsa-miR-21 and mmu-miR-21 with human Smad7 3′-UTR and mouse Smad7 3′-UTR based on TargetScan and Pictar software (http://www.targetscan.org/ and http://pictar.mdcberlin.de/). Several nucleotides in the 50-region of miR-21 (human and mouse) contain a perfect match with the 3′-UTR sequences of the human and mouse Smad7 genes. b The results of luciferase reporter assays. c Representative immunocytochemical images of Smad7 and propidium iodide (PI) staining. Cardiac fibroblasts were treated with TGF-β1 in the presence or absence of miR-21 for 48 h. d Quantitative analysis of Smad7 expression by qRT-PCR. The relative Smad7 mRNA expression ratio (Smad7/GAPDH in arbitrary units) was normalized to that of group CR and the relative expression levels (in fold expression) were calculated. e Representative Western blot depicting the expression of Smad7 protein. f Mean Smad7 protein levels in the control (CR), miR-control (M), pre-miR-21 (PM), TGF-β1 (T), TGF-β1 + pre-miR-21 (TP) and TGF-β1 + miR-21 inhibitor (TI) groups (*p < 0.05 vs. CR, # p < 0.05 vs. T, unpaired t test; n = 10 independent samples for each group)
Discussion
In this study, we examined the relationship between miR-21 and TGF-β1/Smad7 signaling in AF-induced atrial fibrosis. The major findings of this study include the following: (1) our real-time PCR and quantitative Western blot analyses revealed that RAP-induced AF significantly down-regulated Smad7 expression, which was associated with an increase in TGF-β1 and collagen I/III; (2) the AF-induced up-regulation of miR-21 was negatively correlated with Smad7 expression; (3) in isolated adult rat cardiac fibroblasts, treatment with TGF-β1 caused the down-regulation of Smad7 expression and increased the levels of miR-21 and collagen I/III; (4) the luciferase reporter assays suggested that Smad7 is a validated miR-21 target in CFs; and (5) in the presence of TGF-β1, inhibiting or upregulating miR-21 could increase and decrease Smad7 expression, respectively, indicating that the TGF-β1-induced decrease in Smad7 expression might be mediated by miR-21. These results strongly suggest that the miR-21-mediated degradation of Smad7 might decrease the inhibitory feedback regulation of TGF-β1/Smad signaling and serves as a new insight into the development of atrial fibrosis due to atrial fibrillation.
AF and atrial fibrosis
Several animal models of chronic sustained AF have been developed to assess the atrial electric remodeling and arrhythmias of AF. In AF, atrial effective refractory period (AERP) was decreased, and the animals became more vulnerable to AF [4, 34]. It has been reported that rapid pacing induces AF with much higher incidence at the left atrial site than at the right atrium or any other sites, partly because of the inhomogeneous dispersion of AERP. When atrial electrical remodeling returns back to normal, the underlying reasons for persistent AF and structural remodeling are still present. Thus, atrial electrical remodeling alone is sufficient to induce AF, while structural remodeling is responsible for maintaining AF [35, 36]. In this study, we have successfully established the electrical stimulation rapid pacing left atrium-induced AF model. We demonstrated that RAP alone induced profound changes in gene expression of collagens. In addition to the increase in the hydroxyproline content and left atrial weight, a significant deposition of cardiac collagen I and III was observed in the atria subjected to rapid pacing.
TGF-β1 induces myocardial fibrosis by upregulating miR-21
Tachycardia-induced atrial fibrosis is a hallmark of AF-induced structural remodeling. This condition is characterized by the accumulation of extracellular matrix (ECM) and myocardial fibrosis [10, 37]. In this study, we demonstrated that RAP alone induced profound changes in the expression of collagen I/III in the heart, strongly suggesting that tachycardia during AF might cause atrial remodeling resulting from atrial fibrosis. In addition to the increase in hydroxyproline content and left atrial weight, a significant deposition of cardiac collagen I and III was observed in the atria subjected to rapid pacing. These results also support our previous observations that atrial fibrosis consisting of collagen types I and III is a typical feature of AF.
Increasing evidence indicates that targeting TGF-β1/Smad-specific miRNAs related to fibrosis may be a better approach for combating heart disease [38–40], with miR-21 being the best characterized miRNA associated with TGF-β1-mediated fibrosis [41]. It reported that miR-21 is expressed much higher in cardiac fibroblasts and accompanied by the increased expression of collagen in pathological states of heart failure [42]. In a rat model of myocardial infarction, the down-regulation of miR-21 can reduce the degree of atrial fibrosis and the incidence of AF [43]. Adam recently confirmed miR-21 levels were significantly higher in patients with atrial fibrillation, and the up-regulation of miR-21 target genes can increase fibrin deposition [19]. We have observed previously that RAP may cause atrial fibrosis through the AngII/AT1 receptor–specific activation of the TGF-β1/Smad pathway and that TGF-β1 overexpression induces myocardial fibrosis in AF [10]. However, whether miR-21 over-expression enhanced TGF-β1-induced myocardial fibrosis in AF remained elusive. In vivo, we found that RAP induced TGF-β1 and miR-21 up-regulation with the increased expression of collagen. Transfection with the miR-21 inhibitor, we found it reduce the incidence of AF and ameliorated RAP-induced atrial fibrosis. In vitro, we found that miR-21 over-expression increased collagen I/III expression and that miR-21 over-expression after treatment with TGF-β1 further increased collagen I/III expression. In addition, blocking miR-21 expression reverses the TGF-β1-induced up-regulation of collagen I/III. Thus, our study further suggested that miR-21 may be involved in the regulation of RAP-induced myocardial fibrosis, and we speculate that miR-21 over-expression significantly enhances TGF-β1-induced collagen expression.
Mechanisms underlying AF-induced down-regulation of Smad7 expression
The down-regulation of Smad7 may be key in the increased activation of the TGF-β1/Smad signaling pathway that is responsible for increased atrial fibrosis during AF [10, 44]. Smad7 not only exhibits a protective role in Ang II-induced cardiac fibrosis and inflammation, but also possesses therapeutic potential for hypertensive cardiac disease [11, 16]. In our previous study, we reported that the overexpression of Smad7 blocked AngII-induced increases in collagen I synthesis and that the overexpression of TGF-β1 decreased Smad7 expression [10]. In this study, we also found that RAP decreased Smad7 expression, which is negatively correlated with TGF-β1 and collagen I/III expression. This finding further confirmed that the down-regulation of Smad7 might cause myocardial fibrosis in AF. miR-21 is the best characterized miRNA associated with TGF-β1-mediated fibrosis [41]. For example, miR-21 and Smad7 are critical regulators of TGF-β1 signaling during the induction of carcinoma-associated fibroblast formation [33, 45, 46]. miR-21 overexpression enhances the TGF-β1-induced epithelial-to-mesenchymal transition by targeting Smad7 and aggravates renal damage in diabetic nephropathy. However, the regulation mechanism linking miR-21 and Smad7 in RAP-induced myocardial fibrosis was unclear. To further demonstrate our observations in this study and gain more information about how Smad7 is down-regulated during RAP, we examined growth-arrested adult rat fibroblast cells. Moreover, bioinformatics and the luciferase reporter assays showed that Smad7 is a validated target of miR-21. We found that miR-21 over-expression decreases Smad7 expression, which was similar to the change that occurred with TGF-β1 treatment. Furthermore, miR-21 over-expression significantly enhanced TGF-β1-induced Smad7 down-regulation. Interesting, inhibiting miR-21 expression weakened the TGF-β1-mediated down-regulation of Smad7 expression. This study further confirmed our previous research that the down-regulation of Smad7 may be responsible for RAP-induced myocardial fibrosis. More interestingly, the molecular mechanism underlying this phenomenon is as follows: the expression of TGF-β1 leads to miR-21 up-regulation, and miR-21 over-expression directly down-regulates Smad7 expression, which leads to amplification of TGF-β1 signaling and ultimately results in RAP-induced fibrosis. Thus, we speculate that miR-21 over-expression significantly enhances TGF-β1-induced collagen expression and that Smad7 can directly inhibit collagen expression in RAP-induced myocardial fibrosis.
In summary, our data demonstrate that TGF-β1-induced miR-21 over-expression enhances TGF-β1-induced collagens expression by directly down-regulating Smad7. These results may provide new insights into the mechanisms underlying AF-induced atrial fibrosis and myocardial remodeling and provide valuable information for novel therapeutic targets for AF.
References
Burstein B, Nattel S (2008) Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol 51:802–809
Matsuda H, Parwani AS, Attanasio P, Huemer M, Wutzler A, Blaschke F, Haverkamp W, Boldt LH (2015) Atrial rhythm influences catheter tissue contact during radiofrequency catheter ablation of atrial fibrillation: comparison of contact force between sinus rhythm and atrial fibrillation. Heart Vessels. doi:10.1007/s00380-015-0763-0
Tan AY, Zimetbaum P (2011) Atrial fibrillation and atrial fibrosis. J Cardiovasc Pharmacol 57:625–629
Hoshiyama T, Yamabe H, Koyama J, Kanazawa H, Ogawa H (2015) Left atrial electrophysiologic feature specific for the genesis of complex fractionated atrial electrogram during atrial fibrillation. Heart Vessels. doi:10.1007/s00380-015-0672-2
Aldhoon B, Melenovsky V, Peichl P, Kautzner J (2010) New insights into mechanisms of atrial fibrillation. Physiol Res 59:1–12
Csepe TA, Kalyanasundaram A, Hansen BJ, Zhao J, Fedorov VV (2015) Fibrosis: a structural modulator of sinoatrial node physiology and dysfunction. Front Physiol 6:37
de Oliveira IM, Oliveira BD, Scanavacca MI, Gutierrez PS (2013) Fibrosis, myocardial crossings, disconnections, abrupt turns, and epicardial reflections: do they play an actual role in human permanent atrial fibrillation? A controlled necropsy study. Cardiovasc Pathol 22:65–69
Gramley F, Lorenzen J, Koellensperger E, Kettering K, Weiss C, Munzel T (2010) Atrial fibrosis and atrial fibrillation: the role of the TGF-beta1 signaling pathway. Int J Cardiol 143:405–413
Gao X, He X, Luo B, Peng L, Lin J, Zuo Z (2009) Angiotensin II increases collagen I expression via transforming growth factor-beta1 and extracellular signal-regulated kinase in cardiac fibroblasts. Eur J Pharmacol 606:115–120
He X, Gao X, Peng L, Wang S, Zhu Y, Ma H, Lin J, Duan DD (2011) Atrial fibrillation induces myocardial fibrosis through angiotensin II type 1 receptor-specific Arkadia-mediated downregulation of Smad7. Circ Res 108:164–175
Yan X, Liu Z, Chen Y (2009) Regulation of TGF-beta signaling by Smad7. Acta Biochim Biophys Sin (Shanghai) 41:263–272
Meng XM, Tang PM, Li J, Lan HY (2015) TGF-beta/Smad signaling in renal fibrosis. Front Physiol 6:82
Jazbutyte V, Thum T (2010) MicroRNA-21: from cancer to cardiovascular disease. Curr Drug Targets 11:926–935
Yang Y, Yang JJ, Tao H, Jin WS (2015) MicroRNA-21 controls hTERT via PTEN in human colorectal cancer cell proliferation. J Physiol Biochem 71:59–68
Dong S, Ma W, Hao B, Hu F, Yan L, Yan X, Wang Y, Chen Z, Wang Z (2014) MicroRNA-21 promotes cardiac fibrosis and development of heart failure with preserved left ventricular ejection fraction by up-regulating Bcl-2. Int J Clin Exp Pathol 7:565–574
Bhagat TD, Zhou L, Sokol L, Kessel R, Caceres G, Gundabolu K, Tamari R, Gordon S, Mantzaris I, Jodlowski T, Yu Y, Jing X, Polineni R, Bhatia K, Pellagatti A, Boultwood J, Kambhampati S, Steidl U, Stein C, Ju W, Liu G, Kenny P, List A, Bitzer M, Verma A (2013) MiR-21 mediates hematopoietic suppression in MDS by activating TGF-beta signaling. Blood 121:2875–2881
Chung AC, Dong Y, Yang W, Zhong X, Li R, Lan HY (2013) Smad7 suppresses renal fibrosis via altering expression of TGF-beta/Smad3-regulated microRNAs. Mol Ther 21:388–398
Duygu B, Da CMP (2015) miR-21: a star player in cardiac hypertrophy. Cardiovasc Res 105:235–237
Lan HY, Chung AC (2011) Transforming growth factor-beta and Smads. Contrib Nephrol 170:75–82
Bonci D (2010) MicroRNA-21 as therapeutic target in cancer and cardiovascular disease. Recent Pat Cardiovasc Drug Discov 5:156–161
Buscaglia LE, Li Y (2011) Apoptosis and the target genes of microRNA-21. Chin J Cancer 30:371–380
Liang H, Zhang C, Ban T, Liu Y, Mei L, Piao X, Zhao D, Lu Y, Chu W, Yang B (2012) A novel reciprocal loop between microRNA-21 and TGFbetaRIII is involved in cardiac fibrosis. Int J Biochem Cell Biol 44:2152–2160
Orenes-Pinero E, Montoro-Garcia S, Patel JV, Valdes M, Marin F, Lip GY (2013) Role of microRNAs in cardiac remodelling: new insights and future perspectives. Int J Cardiol 167:1651–1659
Garcia R, Nistal JF, Merino D, Price NL, Fernandez-Hernando C, Beaumont J, Gonzalez A, Hurle MA, Villar AV (2015) p-SMAD2/3 and DICER promote pre-miR-21 processing during pressure overload-associated myocardial remodeling. Biochim Biophys Acta 1852:1520–1530
Wang JY, Gao YB, Zhang N, Zou DW, Wang P, Zhu ZY, Li JY, Zhou SN, Wang SC, Wang YY, Yang JK (2014) miR-21 overexpression enhances TGF-beta1-induced epithelial-to-mesenchymal transition by target smad7 and aggravates renal damage in diabetic nephropathy. Mol Cell Endocrinol 392:163–172
Moriguchi M, Niwano S, Yoshizawa N, Kitano Y, Kojima J, Inuo K, Saitou J, Izumi T (2001) Inhomogeneity in the appearance of electrical remodeling during chronic rapid atrial pacing: evaluation of the dispersion of atrial effective refractoriness. Jpn Circ J 65:335–340
Sheeha C, Hrapchak B (1980) Theory and practice of histotechnology. Battelle Press, Columbus, pp 24–36
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−∆∆ C(T)) Method. Methods 25:402–408
Bumrungthai S, Ekalaksananan T, Evans MF, Chopjitt P, Tangsiriwatthana T, Patarapadungkit N, Kleebkaow P, Luanratanakorn S, Kongyingyoes B, Worawichawong S, Pientong C (2015) Up-regulation of miR-21 is associated with cervicitis and human papillomavirus infection in cervical tissues. PLoS ONE 10:e0127109
Rosato P, Anastasiadou E, Garg N, Lenze D, Boccellato F, Vincenti S, Severa M, Coccia EM, Bigi R, Cirone M, Ferretti E, Campese AF, Hummel M, Frati L, Presutti C, Faggioni A, Trivedi P (2012) Differential regulation of miR-21 and miR-146a by Epstein-Barr virus-encoded EBNA2. Leukemia 26:2343–2352
Sheng H (1995) Study on the quantitative methods of immunohistochemistry. Chin J Histochem Cytochem 4:89–92
Kumarswamy R, Volkmann I, Thum T (2011) Regulation and function of miRNA-21 in health and disease. RNA Biol 8:706–713
Li Q, Zhang D, Wang Y, Sun P, Hou X, Larner J, Xiong W, Mi J (2013) MiR-21/Smad 7 signaling determines TGF-beta1-induced CAF formation. Sci Rep 3:2038
Fukaya H, Niwano S, Satoh D, Masaki Y, Niwano H, Kojima J, Moriguchi M, Izumi T (2008) Inhomogenic effect of bepridil on atrial electrical remodeling in a canine rapid atrial stimulation model. Circ J 72:318–326
Anne W, Willems R, Holemans P, Beckers F, Roskams T, Lenaerts I, Ector H, Heidbuchel H (2007) Self-terminating AF depends on electrical remodeling while persistent AF depends on additional structural changes in a rapid atrially paced sheep model. J Mol Cell Cardiol 43:148–158
Cha TJ, Ehrlich JR, Zhang L, Shi YF, Tardif JC, Leung TK, Nattel S (2004) Dissociation between ionic remodeling and ability to sustain atrial fibrillation during recovery from experimental congestive heart failure. Circulation 109:412–418
Kalman JM, Kumar S, Sanders P (2012) Markers of collagen synthesis, atrial fibrosis, and the mechanisms underlying atrial fibrillation. J Am Coll Cardiol 60:1807–1808
Kim GH (2013) MicroRNA regulation of cardiac conduction and arrhythmias. Transl Res 161:381–392
Qin W, Chung AC, Huang XR, Meng XM, Hui DS, Yu CM, Sung JJ, Lan HY (2011) TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J Am Soc Nephrol 22:1462–1474
Velu VK, Ramesh R, Srinivasan AR (2012) Circulating microRNAs as biomarkers in health and disease. J Clin Diagn Res 6:1791–1795
Kato M, Arce L, Wang M, Putta S, Lanting L, Natarajan R (2011) A microRNA circuit mediates transforming growth factor-beta1 autoregulation in renal glomerular mesangial cells. Kidney Int 80:358–368
Cardin S, Guasch E, Luo X, Naud P, Le Quang K, Shi Y, Tardif JC, Comtois P, Nattel S (2012) Role for microRNA-21 in atrial profibrillatory fibrotic remodeling associated with experimental postinfarction heart failure. Circ Arrhythm Electrophysiol 5:1027–1035
Adam O, Lohfelm B, Thum T, Gupta SK, Puhl SL, Schafers HJ, Bohm M, Laufs U (2012) Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res Cardiol 107:278
Wei LH, Huang XR, Zhang Y, Li YQ, Chen HY, Heuchel R, Yan BP, Yu CM, Lan HY (2013) Deficiency of Smad7 enhances cardiac remodeling induced by angiotensin II infusion in a mouse model of hypertension. PLoS ONE 8:e70195
Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, Kaminski N, Abraham E (2010) miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med 207:1589–1597
Zhong X, Chung AC, Chen HY, Dong Y, Meng XM, Li R, Yang W, Hou FF, Lan HY (2013) miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia 56:663–674
Acknowledgments
No fund received for this study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
He, X., Zhang, K., Gao, X. et al. Rapid atrial pacing induces myocardial fibrosis by down-regulating Smad7 via microRNA-21 in rabbit. Heart Vessels 31, 1696–1708 (2016). https://doi.org/10.1007/s00380-016-0808-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00380-016-0808-z