
Abstract
In this study, trivalent rare-earth ion (Eu3+, Gd3+, and Yb3+)-substituted silicate-based bioactive glass scaffolds were prepared by robocasting method using sol–gel-derived bioactive glass powders for tissue engineering applications and cancer therapy. The structural, morphological, and mechanical properties of the prepared scaffolds as well as their in vitro bioactivity in simulated body fluid (SBF) were investigated in detail. In addition, an anticancer drug (5-FU) adsorption and release behavior of the scaffolds was studied as a function of time. In vitro, cytotoxicity and alkaline phosphatase activity were investigated using human skin fibroblast BJ and osteosarcoma SaOS-2 cells. Results showed that using lanthanide ion-containing (0.5, 1, 3, and 5 wt%) sol–gel-derived bioactive glass powders it was possible to successfully fabricate periodic, mesh-like patterned robocast glass scaffolds. All of the scaffolds prepared in the study sintered at 675 °C showed an amorphous structure. The compressive strength of scaffolds was in the range of 8.8 MPa to 13.6 MPa and the highest strength values were obtained in the Yb3+-containing scaffolds. Hydroxyapatite formation was obtained for the scaffolds immersed in SBF for 28 days. The fluorouracil adsorption amount was calculated to be ~ 25% for all types of scaffolds and the cumulative drug release was in the range of 20–25% depending on the dopant concentration. Results of the in vitro cell culture experiments revealed that all of the scaffolds fabricated in the study were not cytotoxic to fibroblast and osteosarcoma cells for up to 7 days under in vitro conditions. An increase was obtained for the ALP activities for both types of cells as the incubation time was increased.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
For the development of innovative and effective methods in cancer treatment, materials science has emerged as a promising frontier. Among the intriguing developments in this field is the utilization of bioactive glasses, a unique class of materials with the potential to revolutionize bone cancer therapy [1, 2]. Bioactive glasses, originally designed for their ability to bond with living tissues and promote tissue regeneration [3,4,5], have garnered significant attention for their remarkable properties as carriers for therapeutic ions [6], particularly rare-earth ions [7,8,9,10,11]. Trivalent rare-earth ions have unique optical, magnetic, and luminescence characteristics that make them invaluable in a wide range of technological and biomedical applications [12]. When embedded within bioactive glasses, these ions open up new avenues for advanced drug delivery, tissue engineering, and diagnostics. The integration of rare-earth elements (REEs) into bioactive glasses not only enhances their biocompatibility but also unlocks a range of powerful functionalities that hold great promise for targeted cancer treatment [13]. Among these elements, europium (III), gadolinium (III), and ytterbium (III) have gained prominence for their magnetic, luminescent, and optical properties [14,15,16]. However, as their usage expands, concerns about their potential cytotoxicity have arisen, prompting researchers to investigate the effects of these elements on living cells.
Photoluminescence and toxicity examination of red emissive Eu3+ complex cell dye has been investigated by Canisares et al.[17]. The cytotoxicity of a Eu3+-containing complex was examined for CHO-K1 cells using an MTT assay, and no obvious toxicity was observed after 24 h of exposure at a concentration in the range of 25–200 µg/mL [17]. Based on the previous study of Andrenuscu et al. [18] about the Eu3+-doped hydroxyapatite nanoparticles, after 5 days of cultivation with stem cells (amniotic fluid), no cytotoxicity was observed.
While gadolinium-based contrast agents (GBCAs) have been considered safe for diagnostic purposes, there have been concerns about their long-term effects and potential cytotoxicity [19]. Research on the cytotoxicity of gadolinium has primarily focused on the stability of GBCAs in the body and the potential for gadolinium deposition in tissues. In some cases, gadolinium deposits have been observed in the brain and other organs, particularly in individuals with impaired kidney function [20]. Toxicity of GBCAs has been attributed to the dissociation of Gd3+ from the chelated complexes [19, 21]. However, the clinical significance of these deposits and their associated cytotoxicity remain subjects of ongoing investigation.
The cytotoxicity of ytterbium, like other rare-earth elements, is largely dependent on its chemical form and concentration. Ytterbium ions can interfere with cellular processes when present in excessive amounts. However, studies have shown that ytterbium-based contrast agents, designed for medical imaging, can be used safely at appropriate concentrations without causing significant cytotoxic effects [22, 23]. A past study by Selvaraj and co-workers [24] demonstrated that the ytterbium oxide nanoparticle exposure at different concentrations (0–50 μg/mL) caused increased cellular apoptosis and necrosis in cultured human kidney HEK293 cells.
Previously, Eu3+, Gd3+, and Yb3+-substituted bioactive glass powders have been prepared through the sol–gel method and the structural, photoluminescence characteristics of the prepared glass powders as well as the hydroxyapatite formation behavior in SBF and its relation with the luminescence and optical oxygen sensitivity characteristics were reported [15, 25]. This study aims to provide an exploration of the structural, morphological, and mechanical properties as well as bioactivity, and cytotoxicity against human osteosarcoma SaOS-2 and skin fibroblast BJ cells, and the anticancer release behavior of trivalent lanthanide element (Eu3+, Gd3+, or Yb3+)-containing 13–93 glass three dimensional periodic scaffolds prepared by robocasting. By shedding light on the multifaceted capabilities of REE-containing bioactive glasses, the study focuses on inspiring further research and driving the development of novel materials for improved healthcare and therapeutic interventions.
2 Experimental studies
2.1 Bioactive glass periodic scaffold manufacture
Three-dimensional bioactive glass scaffolds having periodic grid-like structures were manufactured using sol–gel-derived amorphous bioactive glass powders by the robocasting method (Fisnar NV 7400N, USA). The scaffold manufacturing process involves (1) bioactive glass particle synthesis using a sol–gel process, (2) ink preparation by mixing synthesized glass particles with a temperature-sensitive hydrogel, and (3) scaffold preparation by robocasting (printing) followed by sintering.
The procedure followed for the synthesis of the silicate-based 13–93 bioactive glass (BG) particles containing Eu3+, Gd3+ and Yb3+ at different concentrations (53 SiO2–x, 6 Na2O, 12 K2O, 5 MgO, 20 CaO, 4 P2O5 wt%, where x = 0.5, 1, 3, 5 wt% Eu2O3, Gd2O3, Yb2O3) can be found elsewhere [15, 25]. For the glass synthesis tetraethyl-orthosilicate was dissolved in a water solution containing nitric acid at 25 °C, leading to hydrolysis, followed by agitation for 60 min. Subsequently, a sequence of other chemicals (triethyl-phosphate, calcium nitrate tetrahydrate, magnesium nitrate hexahydrate, sodium nitrate, potassium nitrate, europium(III) nitrate pentahydrate or gadolinium(III) nitrate hexahydrate, or ytterbium(III) nitrate pentahydrate) was added and the mixture was further stirred for 30 min. After gel formation, aging at 60 °C (24 h), and drying at 120 °C (24 h) in an air atmosphere prepared bioactive glass powders were calcined at 625 °C for 4 h using a heating rate of 5 °C/min. Then they were ground using a planetary ball mill (Fritch Pulverisette 7, Premium Line) for 10 min at 700 rpm using Al2O3 balls for size reduction.
The aqueous Pluronic F-127 (Sigma Aldrich) solution (25 wt%) was prepared using a magnetic stirrer at room temperature and the prepared polymer solution was kept at + 4 °C. Then, rare-earth ion-containing sol–gel-derived bioactive glass particles (35 wt%) were mixed with the F-127 solution to prepare the printing ink for robocasting. For this purpose, the mixture was homogenized using a planetary mixer (Uni-Cyclone UM-113, Japan) for 5 min at 1000 rpm. The obtained suspension was well-dispersed and any other dispersant such as polyacrylic acid and another type of block-copolymer [26] that is commonly used in the stabilization of inorganic particles in the liquid-based system was not utilized. The disc-shaped, periodic, mesh-like porous scaffolds with a diameter of 1 cm were printed at room temperature using a nozzle (diameter 510 µm) attached to a dispensing syringe [27]. The disc-shaped as well as cylindrical constructs, with the pore width between the two-rod centers (x–y—direction), were adjusted as 1000 µm. After casting, the scaffolds were sintered at 675 °C for 1 h [28].
2.2 Characterizations
Structural characterizations were performed using an X-ray diffractometer (XRD, Malvern Panalytical brand, Empyrean, CuKα radiation) and a Fourier transform infrared spectrometer (FTIR, Thermo Nicolet IS20,USA) in the presence of ATR accessory (wavelength range 525–4000 cm−1). The morphology of the prepared bioactive glasses was investigated using SEM (Zeiss, Gemini 500, Germany), and an optical microscope (Zeiss, Germany). The surface of the glass samples was sputter-coated by Au/Pd before SEM analysis. The particle size of the glass powders was measured using a Malvern Mastersizer 3000 particle size analyzer. The BET surface area of the synthesized glass particles was measured based on N2 adsorption using Micromeritics Gemini VII 2390. Viscosity and the shear stress of the prepared printing inks containing bare bioactive glass powders (35wt%) and the aqueous Pluronic F-127 hydrogel (25 wt%) were measured as a function of shear rate using a viscometer (Thermo Scientific, Viscotester E) by small sample adaptor (spindle TR11) at 25 °C. The compressive strength of the manufactured bioactive glass multi-layered, cylindrical scaffolds was tested using a mechanical compression testing device (Shimadzu, Japan) with a deformation rate of 0.5 mm/min.
2.2.1 In vitro bioactivity
The conversion of the fabricated bioactive glass scaffolds into hydroxyapatite was investigated in simulated body fluid (SBF) at 37 °C under stationary conditions. The SBF was prepared following the procedure outlined by Kokubo et al. [29], involving the use of NaCl, NaHCO3, KCl, K2HPO4.3H2O, MgCl2.6H2O, CaCl2, Na2SO4, and NH2C(CH2OH)3. The bioactive glass scaffolds were submerged in SBF with an initial pH of 7.4 (1 g of sample per 500 ml of SBF) and incubated in an incubator for up to 28 days. Subsequently, they were extracted from the SBF, dried at 60 °C for 24 h, and subjected to analysis. The presence of hydroxyapatite on the surface of the SBF-treated samples was evaluated using SEM and FTIR techniques under previously defined conditions.
2.2.2 Drug delivery
Fluorouracil (5-FU, Sigma Aldrich) was used as the model drug in the study. For the drug loading experiments, each scaffold was immersed in 10 ml of drug solution and kept under dark conditions at 25 °C for 48 h. A UV–Vis spectrophotometer (Thermo-Scientific, Evolution 201, USA) was used to determine the drug concentration and amount of drug adsorbed onto the glass samples at 266 nm. The bioactive glass samples were dried at 40 °C for 48 h after being separated from the drug solution. Samples with known weight and loaded drug amount were stored in phosphate-buffered saline solution (PBS, pH 7.4) at 37 °C for drug release tests. During experiments a 2 ml sample was taken and replaced with a 2 ml fresh solution until the drug release plateau was reached. To observe any burst effect, the initial sample collection was done at tighter intervals. Samples were taken at specific time intervals (hourly for the first 24 h and up to 320 h), and absorbance values at 266 nm were determined using a UV–Vis spectrophotometer.
2.2.3 In vitro cell culture experiments
In the study, human osteosarcoma SaOS-2 cells (ATCC, HTB-85), and human fibroblast BJ (ATCC, CRL-2522) cells were utilized in cell culture experiments. It has been reported that SaOS-2 cells have epithelial morphology and were isolated from the bone of an osteosarcoma patient [30, 31]. On the other hand, BJ fibroblasts are isolated from human normal skin [32].
In 75 cm2 culture dishes, the SaOS-2 cells were grown in McCoy’s 5A medium (Modified, Sigma-Aldrich) with 10% fetal bovine serum (FBS, Sigma-Aldrich), 2 mM glutamine and 1% penicillin–streptomycin (Sigma-Aldrich) at 37 °C with 5% CO2 and 96% humidity. While BJ fibroblast cells were cultured in DMEM (Sigma-Aldrich) with 10% FBS and antibiotics (1% Penicillin–Streptomycin, Sigma Aldrich). When the cells were 70–80% confluent, they were removed from the flask using a 0.05% trypsin/0.02 EDTA (Gibco, USA) solution, suspended in growth media, and centrifuged for 5 min at 1500 rpm. The cells were seeded into well plates at a density of 5 × 104 cells/well after the supernatant had been removed. In the absence of the bioactive glasses, the cells in the culture medium were used as the control group. The cells were incubated for 1, 3, and 7 days by changing the cell culture medium every 2 days during the experiments.
2.2.3.1 Cytotoxicity
To assess cell viability, the MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) which is a colorimetric test used to assess cell metabolic activity (Sigma-Aldrich) was performed [33].
Before seeding cells on the bioactive glass in well plates, samples were sterilized using a high-temperature furnace (350 °C, 5 h) followed by immersion in 70% ethanol overnight and exposure to UV light for 1 h. Bioactive glass scaffolds were immersed in separate wells using a 24-well plate. Totally 5 × 104 cells were seeded in 100 μl of medium per well, allowing the cells to distribute within the glass samples. For the MTT analysis, the culture medium in each well of the culture plate was removed at specific time points, and 600 μl of serum-free culture medium and 60 μl of MTT solution (2.5 mg/ml in PBS) were added to each well. After incubating at 37 °C for 4 h, the medium containing MTT was removed, and dimethyl sulfoxide (DMSO, Sigma-Aldrich) was added to each well to dissolve the resulting purple formazan crystals. The absorbance of the obtained purple-colored solution was measured at 570 nm using a microplate reader. For each condition, three replicate samples were examined.
2.2.3.2 ALP activity
Alkaline phosphatase activity (ALP) of the cell-seeded bioactive glass samples was measured using an Alkaline Phosphatase Activity Fluorometric Assay Kit (Sigma-Aldrich, MAK411) based on the procedure provided by the manufacturer. In the kit, ALP splits the phosphate group of the non-fluorescent 4-methylumbelliferyl phosphate disodium salt (4-MUP) substrate arising in a strong fluorescent signal (λEx = 360 nm and λEm = 440 nm). ALP activity of the samples was measured after 1, 3, and 7 days of culture.
2.3 Statistical analysis
The student’s t test or one-way ANOVA followed by Tukey’s post hoc test was performed. Each experiment was performed in triplicate and the data were presented as mean ± standard deviation. The p value < 0.05 (*) or 0.01 (**) and 0.001 (***) were considered significant.
3 Results and discussion
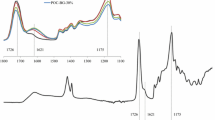
The structural and morphological properties of the synthesized pristine silicate-based bioactive glass particles are given in Fig. 1a–e. FTIR spectrum of the 13–93 glass gel dried at 120 °C (24 h) and the powder calcined at 625 °C (4 h) are shown in Fig. 1a, b. In IR spectra, the band in the range of 980–1100 cm−1 is related to the asymmetric stretching of Si–O–Si, where oxygen atoms move horizontally within the Si–O–Si plane [34, 35]. The band between 1350 and 1400 cm−1 can be attributed to the presence of NO3− ions within the glass network and it becomes very weak for the calcined powder [36]. In addition, the -OH band shown in the IR spectrum of the dried gel at around 3500 cm−1 does not exist in the spectrum of the powder calcined at 625 °C.

FTIR spectrum of the bare 13–93 bioactive glass gel a after drying at 120 °C for 24 h, b after calcination at 625 °C for 4 h; c particle size distribution graph, d BET surface area plot, e SEM micrograph of the synthesized 13–93 glass powders after size reduction. Inset image: photograph of the prepared bioactive glass gels before heat treatment
It is seen that the synthesized bare silicate-based bioactive glass powders have a wide particle size distribution. According to the measurement results made with the particle size analyzer, it is understood that the d50 particle size of the bare, 5Eu3+, 5Gd3+, and 5Yb3+-substituted silicate glass powders was ~ 3.30 µm, ~ 2.70 µm, ~ 3.17 µm, and ~ 3.59 µm, respectively. SEM micrograph of the bare 13–93 bioactive glass powder also shows the presence of some sub-micron-sized particles. The BET surface area of the bare 13–93 glass powder was measured to be 2.168 m2/g.
The flow curve of the printing ink containing bare 13–93 bioactive glass powders (35 wt%) and the aqueous F-127 hydrogel solution is shown in Fig. 2a. An image of the printing ink before the robocasting process is also given in Fig. 2b. Accordingly, the bioactive glass dispersion showed a shear thinning behavior which is a requirement for a successful printing operation. In addition, rare-earth element -containing bioactive glass dispersions demonstrated a similar rheological behavior (results are not shown).

a Flow curve and b digital image of the printing ink prepared using bare 13–93 bioactive glass powders (35 wt%) and the F-127 hydrogel (25 wt%) mixture
Optical microscope images of the bare 13–93 bioactive glass scaffolds fabricated using sol–gel-derived glass particles and sintered at 675 °C are shown in Fig. 3. Scaffolds have oriented porous structure having a strut diameter of approximately 400 ± 100 µm. The diameter and the height of the scaffolds were 8 mm and 5 mm, respectively, regardless of the rare-earth element type and concentration. The pore size and the total porosity of the fabricated bioactive glass scaffolds were around 600 µm and 65–70%, respectively. Figure 4 demonstrates the high-magnification optical microscope images of the prepared scaffolds containing Eu3+, Gd3+, and Yb3+ at different concentrations. Results revealed that the inclusion of the rare-earth elements did not significantly influence the morphological characteristics of the scaffolds. Higher strut diameters obtained for some of the rare-earth element-doped scaffolds may be attributed to the release of the excess trapped nitrates in the bioactive glass structures that occurred during the sintering process. FTIR spectra of the fabricated bioactive glass scaffolds shown in Fig. 5 indicated the presence of the Si–O vibration bands. Specifically, the band around ~ 790 cm−1 corresponds to symmetric stretching within the Si–O–Si plane, where oxygen atoms undergo vertical movement. Moreover, the more pronounced band in the range of 980–1100 cm−1 is related to the asymmetric stretching of Si–O–Si, where oxygen atoms move horizontally within the Si–O–Si plane [34, 35]. The weaker band between 1350 and 1400 cm−1 can be attributed to the presence of NO3− ions within the glass network [36].

Optical microscope images of the bare 13–93 bioactive glass scaffolds fabricated in the study; a, b top view, c side view

Optical microscope images of a Eu3+, b Gd3+, and c Yb3+-containing bioactive glass scaffolds. Scale bar: 500 µm

FTIR spectra of the sintered a Eu3+, b Gd3+, and c Yb3+-containing bioactive glass scaffolds; d XRD pattern of the sintered scaffolds containing RE3+ at highest concentration
It has been reported that by adding REEs to the silicate glass network different properties such as hardness, elastic modulus, and corrosion resistance can be greatly improved since the REE cations have higher field strength than conventional network modifier cations [37,38,39].
In the current study, the influence of the high field strength of REE ions on the stretching vibration of the Si–O–Si bond may be the reason that the absorption peak (located at ~ 920 cm−1) moves to a higher wavenumber. The broadening of the absorption band indicates that the Qn (where Q refers to the degree of polymerization of the glass, and n refers to the number of bridging oxygen) structural unit in the glass network is increased [38].
Based on the XRD patterns given in Fig. 5d, all of the bioactive glass scaffolds fabricated in the study were amorphous after sintering at 675 °C. A decrease in the intensity of the band located at ∼920 cm-1 was obtained as the rare-earth element concentration in the glass network was increased, since during particle synthesis, REE substitution was performed for SiO2.
Results of the mechanical tests showed that the compressive strength of the bare 13–93 bioactive glass scaffolds (8 mm diameter and 10 mm height) fabricated using sol–gel-derived bioactive glass powders was 10.2 ± 1.03 MPa. The inclusion of Eu3+ and Gd3+ at 5% did not cause a significant change in the mechanical properties. A slightly higher compressive strength (13.6 ± 0.9 MPa) was obtained in the case of 5% Yb3+-doped bioactive glass scaffolds (see Fig. 6). The increase obtained in the mechanical properties of the Yb3+-doped bioactive glass scaffolds may be attributed to the role of the Yb3+ ions as a network modifier in the silica glass network and the decrease in the number of the non-bridging oxygens. In general, the compressive strength of the scaffolds was sufficient to use in the repair of trabecular bone defects [40].

Compressive strength of the RE3+-containing (5 wt%) bioactive glass scaffolds
FTIR spectra of the fabricated bioactive glass scaffolds after immersion in simulated body fluid for 7, 14, and 28 days are shown in Fig. 7. In the spectra of all of the glass scaffolds containing Eu3+, Gd3+, and Yb3+ treated in SBF starting from 7 days, a broad band near 1035 cm−1 and doublet peaks at 603 cm−1 and 556 cm−1 have been observed. The ~ 1035 cm−1 band corresponds to the ν3 bending mode of PO43−, while the minor peak at 961 cm−1 is attributed to the ν1 mode of PO43−, and the peaks at ~ 556 cm−1 and 603 cm−1 signify the ν4 P–O bending of PO43−, indicating the presence of orthophosphate lattices [41, 42]. Peaks at ~ 1390 cm−1 and ~ 881 cm−1 are due to the asymmetric stretching (ν3) and out-of-plane bending (ν2) vibrations, respectively, suggesting the formation of carbonates on the glass surface. The ν1 stretching modes of CO32− are represented by the peak at ~ 1480 cm−1 [43]. Results also showed that as the immersion time in SBF increased an increase in band intensities was recorded. The highest intensities for the peaks at ~ 556 cm−1 and 603 cm−1 representing the crystalline HA formation were obtained in samples treated in simulated body fluid for 28 days.

FTIR spectra of a Eu3+, b Gd3+, and c Yb3+-containing bioactive glass scaffolds immersed in SBF for 7, 14 and 28 days
SEM micrographs given in Fig. 8 also demonstrate the deposition of another material having plate-like morphology on the surface of the glass scaffolds after being treated in SBF for 7, 14, and 28 days. The morphology of this second-phase material was well-matched with the hydroxyapatite [44]. SEM micrographs revealed that after immersion in simulated body fluid for 28 days some clusters of HA formed on the surface. The size of the HA crystals grew with the increase in immersion time and the formation of a thick HA layer was detected (see inset plot-crack occurred in HA layer due to increased thickness).

SEM micrographs of the a , b, c 5 Eu3+; d, e, f 5 Gd3+; g, h, i 5 Yb3+-containing bioactive glass robocast scaffolds immersed in SBF for 7, 14, and 28 days
Figure 9 depicts the fluorouracil adsorption percentages of the fabricated bioactive scaffolds that were kept in drug solution for 48 h in a dark environment. Accordingly, the total drug adsorption amount of the scaffolds was in the range of 25–30%. Results of the release experiments showed that the cumulative 5-FU release amount into PBS at pH 7.4 was between 20% and 25% after 300 h (Fig. 10). In the study drug release kinetics were studied using zero order, first order, and Higuchi kinetic models [45]. Results of the drug release kinetic studies showed that 5-FU delivery from the fabricated bioactive glass scaffolds was best fitted by the Higuchi kinetic model. Figure 11 demonstrates the drug release data fitted with the Higuchi model. The coefficient of regression values was in the range of 0.82–0.95 based on this model. Kinetic model parameters of three models calculated for all of the glass compositions prepared in the study are given in Table 1.

a Calibration curve of the 5-FU solution, drug adsorption percentages of the b Eu3+, c Gd3+, and d Yb3+-containing bioactive glass scaffolds

Cumulative drug release percentages of a Eu3+, b Gd3+, and c Yb3+-containing bioactive glass scaffolds in PBS medium at pH 7.4 at 25 °C

Graphs showing the drug release from a bare, b 5%Eu3+, c 5%Gd3+, and d 5% Yb3+-containing bioactive glass scaffolds fitted using Higuchi kinetic model
In vitro biocompatibility of the fabricated bioactive glass scaffolds were investigated using the MTT assay and the ALP activity tests. Figure 12 demonstrates the BJ human fibroblast cell viability percentages and their ALP activities after culturing for 1, 3, and 7 days in the presence of bioactive glass scaffolds containing trivalent REE ions at different concentrations. Accordingly, cell viability percentages were higher than 80% for all of the samples studied after culturing 1 and 3 days. A decrease was obtained at the highest REE concentration after 7 days of culture. It has been known that there is a close relation between cytotoxicity and reactive oxygen species (ROS) generation capacity. Therefore, in the current study, in vitro cytotoxicity observed for some conditions may be related to the generation of reactive oxygen species as well as oxidative stress produced by the bioactive glass samples [46]. The results of the past study of Andronescu et al. [18] showed that the viability of HGF-1 fibroblast was decreased in the Eu3+-containing (10%) hydroxyapatite after 48 h.

Cell viability percentages (left column) and the ALP activities (right column) of the fabricated robocast bioactive glass scaffolds incubated with human skin BJ cells for a 1, b 3, c 7 days
Findings of the current study also showed that BJ fibroblast cells incubated in the presence of bioactive glass scaffolds induced ALP activity. In general, the ALP activity of REE-containing bioactive glass scaffolds was higher than the control group samples (bare 13–93) after 1 and 3 days of culture; however, a decrease in ALP activity was obtained at 5%REE-substituted samples after 7 days which is consistent with the MTT test results. Findings also showed that an increase in ALP activity of the BJ cells was measured as the culture time increased. ALP activity is generally utilized as a marker of osteoblast differentiation and it is an enzyme that byproduct of osteoblast activity; therefore, the high level of ALP activity designates to new bone formation [47].
Results of the in vitro biocompatibility experiments (Fig. 13) which were performed using osteosarcoma SaOS-2 cells showed that studied bioactive glass scaffolds did not cause a cytotoxic response in cells. A slight decrease in cell viability rates was recorded for the 5% Yb3+-containing glass samples after 7 days of incubation. Similar to the BJ fibroblast cells, an increase in ALP activity was obtained for the SaOS-2 cells as the incubation time was extended. Murray et al. [48] reported that the basal ALP activity of SaOS-2 cells was a hundred to thousand times higher than the other human osteogenic sarcoma cell lines. They concluded that the SaOS-2 cell line is an osteoblastic cell model that expresses high levels of tissue-unspecific ALP activity. A past study by Wang et al. [49] indicated that bioactive glass incorporated into the alginate/gelatin hydrogel system improved the proliferation and mineralization of bio-printed osteosarcoma SaOS-2 cells. In the previous studies, SaOS-2 cells have predominantly been employed to assess the biocompatibility of three-dimensional 45S5 bioactive glass scaffolds [50], and mesoporous bioactive glasses [51, 52]. Nonetheless, a past study by Wilkesmann et al. [53] identified the SaOS-2 as the most susceptible cell line in terms of cytotoxicity when exposed to 45S5 bioactive glasses compared to the MG-63 cells. Even at relatively low concentrations of 0.125 mg/ml, SaOS-2 cell viability plummeted below 15% on the 3rd and 6% on the 7th day. Therefore, they concluded that due to the obtained sensitivity of osteoblast-like SaOS-2, HOS, and U2OS cells to bioactive glass samples that cause toxicity, they may not be appropriate to examine the bioactive glasses on osteogenic differentiation and cell viability [53].

Cell viability percentages (left column) and the ALP activities (right column) of the fabricated robocast bioactive glass scaffolds incubated with human osteosarcoma SaOS-2 cells for a 1, b 3, c 7 days
Figures 14 and 15 show the optical microscope images of the BJ fibroblast and the SaOS-2 cells, respectively, incubated for 7 days in the presence of bioactive glass scaffolds fabricated in the current study. A significant difference was not observed in BJ cell densities as a function of rare-earth ion type and concentration; however, a decrease in SaOS-2 cell density was observed for the samples incubated with the Yb3+-doped (especially at high concentration) bioactive glass scaffolds. A previous publication by Zhou and co-workers [46] demonstrated that the existence of yttrium oxide nanoparticles caused a size and concentration-dependent cytotoxicity in primary osteoblastic cells (obtained from mice), and ROS generation may be a damaged pathway for the toxicological effects.

Optical microscope images of the BJ fibroblast cells incubated in the presence of a Eu3+, b Gd3+, and c Yb3+-containing bioactive glass scaffolds for 7 days

Optical microscope images of the SaOS-2 cells incubated in the presence of a Eu3+, b Gd3+, and c Yb3+-containing bioactive glass scaffolds for 7 days
4 Conclusions
In the study, trivalent rare-earth element (Eu3+, Gd3+, and Yb3+) containing (0.5, 1, 3, and 5%) bioactive glass three-dimensional, disc-shaped macroporous scaffolds were fabricated through layer-by-layer assembly using sol–gel-derived bioactive glass powders. Structural characterizations showed that fabricated periodic and mesh-like scaffolds were amorphous after sintering performed at 675 °C. In vitro bioactivity experiments revealed that all of the glass scaffolds fabricated in the study were bioactive and HA formation occurred after immersion in simulated body fluid starting from 7 days. An anticancer agent, 5-FU, adsorption onto the glass surface, and the release at pH 7.4 to the PBS medium was in the range of 25–30% and 20–25%, respectively. A burst drug release was subsequently followed by a sustained fluorouracil delivery behavior was obtained and the release data was best fitted with the Higuchi kinetic model. The cytotoxicity and ALP experiments showed that human skin fibroblast cells did not show any toxic response (except for samples containing REE at the highest concentration) to the studied scaffolds for up to 7 days and they induced ALP activity. Similarly, prepared glass samples, in general, did not cause a decrease in viability percentages of the human osteosarcoma SaOS-2 cells. An increase in ALP activity of the cells was recorded as the incubation time was increased. It was concluded that prepared periodic assemblies made from specific trivalent REE-containing silicate-based bioactive glasses have the potential to be used in cancer therapy.
Data availability
The data sets generated during and/or analyzed throughout the current study are available from the corresponding author upon request.
References
E. Sharifi, A. Bigham, S. Yousefiasl, M. Trovato, M. Ghomi, Y. Esmaeili, P. Samadi, A. Zarrabi, M. Ashrafizadeh, S. Sharifi, R. Sartorius, F. Dabbagh, A. Maleki, H. Song, T. Agarwal, T.K. Maiti, N. Nikfarjam, C. Burvill, V. Mattoli, M.G. Raucci, K. Zheng, A.R. Boccaccini, L. Ambrosio, P. Makvandi, Mesoporous bioactive glasses in cancer diagnosis and therapy: stimuli-responsive, toxicity, immunogenicity, and clinical translation. Adv. Sci. 9, 2102678 (2022). https://doi.org/10.1002/advs.202102678
Y. Lin, J.C. Mauro, G. Kaur, Chapter 10—bioactive glasses for cancer therapy, in Woodhead publishing series in biomaterials, biomedical, therapeutic and clinical applications of bioactive Glasses. ed. by G. Kaur (Woodhead Publishing, 2019), pp.273–312. https://doi.org/10.1016/B978-0-08-102196-5.00010-0
L.L. Hench, R.J. Splinter, W.C. Allen, T.K. Greenlee, Bonding mechanism at the interface of ceramic prosthetic materials. J. Biomed. Mater. Res. 2, 117–141 (1971). https://doi.org/10.1002/jbm.820050611
J.R. Jones, Review of bioactive glass: from Hench to hybrids. Acta Biomater. 9(1), 4457–4486 (2013). https://doi.org/10.1016/j.actbio.2012.08.023. (Epub 2012 Aug 21 PMID: 22922331)
M.N. Rahaman, D.E. Day, B.S. Bal, Q. Fu, S.B. Jung, L.F. Bonewald, A.P. Tomsia, Bioactive glass in tissue engineering. Acta Biomater. 7, 2355-2373 (2011). https://doi.org/10.1016/j.actbio.2011.03.016
V. Mouriño, J.P. Cattalini, A.R. Boccaccini, Metallic ions as therapeutic agents in tissue engineering scaffolds: an overview of their biological applications and strategies for new developments. J. R. Soc. Interface. 9(68), 401–419 (2012). https://doi.org/10.1098/rsif.2011.0611. (Epub 2011 Dec 7. PMID: 22158843; PMCID: PMC3262432)
B.A.E. Ben-Arfa, I.E. Palamá, I.M. Miranda Salvado, J.M.F. Ferreira, R.C. Pullar, Cytotoxicity and bioactivity assessments for Cu2+ and La3+ doped high-silica sol-gel derived bioglasses: the complex interplay between additive ions revealed. J. Biomed. Mater. Res. 107A, 2680–2693 (2019). https://doi.org/10.1002/jbm.a.36772
M. Wang, G. Pi, Bo. Lei, Europium doped monodispersed bioactive glass nanoparticles regulate the osteogenic differentiation of human marrow mesenchymal stem cells. J. Biomed. Nanotechnol. 14, 756–764 (2018). https://doi.org/10.1166/jbn.2018.2504
A.M. Deliormanlı, B. Rahman, S. Oguzlar, M.Z. Ongun, Investigation of the structural, photoluminescence properties, bioactivity and 5-fluorouracil delivery behavior of Dy3+ and Dy:Eu3+-doped bioactive glasses. J. Alloys Compounds. 944, 169153 (2023). https://doi.org/10.1016/j.jallcom.2023.169153
A.M. Deliormanli, S.A.M. Issa, M.S. Al-Buriahi, B. Rahman, H.M.H. Zakaly, H.O. Tekin, Erbium (III)- and Terbium (III)-containing silicate-based bioactive glass powders: physical, structural and nuclear radiation shielding characteristics. Appl. Phys. A 127, 463 (2021). https://doi.org/10.1007/s00339-021-04615-5
A.M. Deliormanlı, M. Türk, Flow behavior and drug release study of injectable pluronic F-127 hydrogels containing bioactive glass and carbon-based nanopowders. J. Inorg. Organomet. Polym. 30, 1184–1196 (2020). https://doi.org/10.1007/s10904-019-01346-2
M. Duchna, I. Cieślik, Rare earth elements in new advanced engineering applications. Rare earth elements—emerging advances technology utilization and resource procurement (IntechOpen, 2023). https://doi.org/10.5772/intechopen.109248
A. Baranowska, M. Kochanowicz, J. Żmojda, P. Miluski, A. Wajda, M. Leśniak, D. Dorosz, Biological properties of rare-earth doped bioactive glass (SPIE, 2020). https://doi.org/10.1117/12.2566347
F. Fernández, R. Sáez-Puche, C. Cascales, C.M. Marcano, I. Rasines, X-ray diffraction data and magnetic properties of the oxides R3Sb5O12 (R = Pr, Nd, Sm, Eu, Gd, Yb). J. Phys. Chem. Solids 50(9), 871–875 (1989). https://doi.org/10.1016/0022-3697(89)90033-4
A.M. Deliormanlı, S.Z. Oguzlar, M. Ongun, Effects of Eu3+, Gd3+ and Yb3+ substitution on the structural, photoluminescence, and decay properties of silicate-based bioactive glass powders. J. Mater. Res. 37, 622–635 (2022). https://doi.org/10.1557/s43578-021-00461-6
N.L. Ignjatović, L. Mančić, M. Vuković, Z. Stojanović, M.G. Nikolić, S. Škapin, S. Jovanović, L. Veselinović, V. Uskoković, S. Lazić, S. Marković, M.M. Lazarević, D.P. Uskoković, Rare-earth (Gd3+, Yb3+/Tm3+, Eu3+) co-doped hydroxyapatite as magnetic, up-conversion and down-conversion materials for multimodal imaging. Sci. Rep. 9(1), 16305 (2019). https://doi.org/10.1038/s41598-019-52885-0
F.S. Canisares, A.M. Mutti, D.G. Cavalcante, A.E. Job, A.M. Pires, S.A. Lima, Luminescence and cytotoxic study of red emissive europium(III) complex as a cell dye. J. Photochem. Photobiol. A Chem. 422, 113552 (2022). https://doi.org/10.1016/j.jphotochem.2021.113552
E. Andronescu, D. Predoi, I.A. Neacsu, A.V. Paduraru, A.M. Musuc, R. Trusca, O. Oprea, E. Tanasa, O.R. Vasile, A.I. Nicoara et al., Photoluminescent hydroxylapatite: Eu3+ doping effect on biological behaviour. Nanomaterials 9(9), 1187 (2019). https://doi.org/10.3390/nano9091187
M. Rogosnitzky, S. Branch, Gadolinium-based contrast agent toxicity: a review of known and proposed mechanisms. Biometals 29(3), 365–376 (2016). https://doi.org/10.1007/s10534-016-9931-7
M.A. Sieber, H. Pietsch, J. Walter, W. Haider, T. Frenzel, H.J. Weinmann, A preclinical study to investigate the development of nephrogenic systemic fibrosis: a possible role for gadolinium-based contrast media. Investig Radiol. 43, 65–75 (2008). https://doi.org/10.1097/RLI.0b013e31815e6277
M. Port, J.M. Idee, C. Medina, C. Robic, M. Sabatou, C. Corot, Efficiency, thermodynamic and kinetic stability of marketed gadolinium chelates and their possible clinical consequences: a critical review. Biometals 21, 469–490 (2008). https://doi.org/10.1007/s10534-008-9135-x
Y.C. Dong, A. Kumar, D.N. Rosario-Berríos, S. Si-Mohamed, J.C. Hsu, L.M. Nieves, P. Douek, P.B. Noël, D.P. Cormode, Ytterbium nanoparticle contrast agents for conventional and spectral photon-counting CT and their applications for hydrogel imaging. ACS Appl. Mater. Interfaces 14(34), 39274–39284 (2022). https://doi.org/10.1021/acsami.2c12354
Z. Liu, Z. Li, J. Liu, Gu. Song, Q. Yuan, J. Ren, Qu. Xiaogang, Long-circulating Er3+-doped Yb2O3 up-conversion nanoparticle as an in vivo X-Ray CT imaging contrast agent. Biomaterials 33(28), 6748–6757 (2012). https://doi.org/10.1016/j.biomaterials.2012.06.033
V. Selvaraj, S. Bodapati, E. Murray, K.M. Rice, N. Winston, T. Shokuhfar, Y. Zhao, E. Blough, Cytotoxicity and genotoxicity caused by yttrium oxide nanoparticles in HEK293 cells. Int. J. Nanomed. 12(9), 1379–1391 (2014). https://doi.org/10.2147/IJN.S52625
S. Oğuzlar, M.Z. Ongun, A.M. Deliormanlı, Modulation of optical oxygen sensitivity of H2(TPP) by using Eu3+, Gd3+ and Yb3+ ions doped silicate-based bioactive glass powders immersed in simulated body fluid. Optik, 294, 171458 (2023). https://doi.org/10.1016/j.ijleo.2023.171458
A. Şakar-Deliormanlı, E. Celik, M. Polat, Adsorption of anionic polyelectrolyte and comb polymers onto lead magnesium niobate. Colloids Surfaces A 316(1–3), 202–209 (2008). https://doi.org/10.1016/j.colsurfa.2007.09.004. (ISSN 0927-7757)
A.M. Deliormanlı, M. Türk, H. Atmaca, Response of mouse bone marrow mesenchymal stem cells to graphene-containing grid-like bioactive glass scaffolds produced by robocasting. J. Biomater. Appl. 33(4), 488–500 (2018). https://doi.org/10.1177/0885328218799610
A.M. Deliormanlı, B. Rahman, Fabrication and characterization of novel multifunctional superparamagnetic and fluorescent bioactive glasses for biomedical applications. J. Alloys Compounds 967, 171653 (2023). https://doi.org/10.1016/j.jallcom.2023.171653
T. Kokubo, H. Takadama, How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 27(15), 2907–2915 (2006). https://doi.org/10.1016/j.biomaterials.2006.01.017
E. Murray, D. Provvedini, D. Curran, B. Catherwood, H. Sussman, S. Manolagas, Characterization of a human osteoblastic osteosarcoma cell line (SAOS-2) with high bone alkaline phosphatase activity. J. Bone Mineral Res. 2, 231–238 (2009). https://doi.org/10.1002/jbmr.5650020310
H.-J. Hausser, R.E. Brenner, Phenotypic instability of Saos-2 cells in long-term culture. Biochem. Biophys. Res. Commun. 333(1), 216–222 (2005). https://doi.org/10.1016/j.bbrc.2005.05.097
S.P. Pereira, C.M. Deus, T.L. Serafim, T. Cunha-Oliveira, P.J. Oliveira, Metabolic and phenotypic characterization of human skin fibroblasts after forcing oxidative capacity. Toxicol. Sci. 164(1), 91–204 (2018). https://doi.org/10.1093/toxsci/kfy068
P.W. Sylvester, Optimization of the tetrazolium dye (MTT) colorimetric assay for cellular growth and viability. Methods Mol. Biol. 716, 157–168 (2011). https://doi.org/10.1007/978-1-61779-012-6_9
A.M. Efimov, V.G. Pogareva, IR absorption spectra of vitreous silica and silicate glasses: the nature of bands in the 1300 to 5000 cm-1 region. Chem. Geol. 229, 198–217 (2006). https://doi.org/10.1016/j.chemgeo.2006.01.022
W. R. Taylor, Application of infrared spectroscopy to studies of silicate glass structure: Examples from the melilite glasses and the systems Na20-SiO2 and Na20-AI203-SiO2, Proc. Indian Acad. Sci. (Earth Planet. Sci.), 99(1), 99–117 (1990)
M. Trivedi, A. Branton, D. Trivedi, G. Nayak, K. Bairwa, S. Jana, Spectroscopic characterization of disodium hydrogen orthophosphate and sodium nitrate after biofield treatment. J. Chromatogr. Sep. Tech. 6(5),1000282 (2015). https://doi.org/10.4172/2157-7064.1000282
A. Aronne, S. Esposito, P. Pernice, FTIR and DTA study of lanthanum aluminosilicate glasses. Mater. Chem. Phys. 51, 163–168 (1997). https://doi.org/10.1016/S0254-0584(97)80287-8
G. Hou, C. Zhang, Fu. Wenbin, G. Li, J. Xia, Y. Ping, Improvement of mechanical strength in Y3+/La3+ co-doped silicate glasses for display screen. Ceram. Int. 45(9), 11850–11855 (2019). https://doi.org/10.1016/j.ceramint.2019.03.066
M.T. Wang, M. Li, J.S. Cheng, F. He, Z.G. Liu, Y.H. Hu, The role of Gd2O3 and Y2O3 in corrosion of soda lime silicate glass. J. Nucl. Mater. 433(1–3), 287–296 (2013). https://doi.org/10.1016/j.jnucmat.2012.09.032
C.E. Misch, Z. Qu, M.W. Bidez, Mechanical properties of trabecular bone in the human mandible: implications for dental implant treatment planning and surgical placement. J. Oral Maxillofac. Surg. 57(6), 700–706 (1999). https://doi.org/10.1016/s0278-2391(99)90437-8
O.H. Andersson, K.H. Karlsson, K. Kngasniemi, Calcium phosphate formation at the surface of bioactive glass in vivo. J. Non Cryst. Solids 119, 290–296 (1990). https://doi.org/10.1016/0022-3093(90)90301-2
G.A. Stanciu, I. Sandulescu, B. Savu, S.G. Stanciu, K.M. Paraskevopoulos, X. Chatzistavrou, E. Kontonasaki, P. Koidis, Investigation of the hydroxyapatite growth on bioactive glass surface. J. Biomed. Pharm. Eng. 1(1), 34–39 (2007)
L. Berzina-Cimdina, N. Borodajenko, Research of calcium phosphates using fourier transform infrared spectroscopy, in Infrared spectroscopy—materials science, engineering and technology. ed. by T. Theophile (InTech, 2012)
S. Gallinetti, C. Canal, M.-P. Ginebra, Development and Characterization of Biphasic Hydroxyapatite/β-TCP Cements. J. Am. Ceram. Soc. 97, 1065–1073 (2014). https://doi.org/10.1111/jace.12861
R.R. Earle, K.K. Bandaru, L. Usha, Formulation and characterization of sustained release coated matrix granules of metformin hydrochloride. Asian J. Pharm. Clin. Res. 11, 387 (2018). https://doi.org/10.22159/ajpcr.2018.v11i7.24996
G. Zhou, Y. Li, Y. Ma, Z.Liu, L.Cao, D.Wang, S.Liu, W. Xu, W. Wang, Size-dependent cytotoxicity of yttrium oxide nanoparticles on primary osteoblasts in vitro. J. Nanopart. Res. 18, 135 (2016). https://doi.org/10.1007/s11051-016-3447-5
M. Ehlert, A. Radtke, T. Jędrzejewski, K. Roszek, M. Bartmanski, P. Piszczek, In vitro studies on nanoporous, nanotubular and nanosponge-like titania coatings, with the use of adipose-derived stem cells. Materials. 13, 1574 (2020). https://doi.org/10.3390/ma13071574
E. Murray, D. Provvedini, D. Curran, B. Catherwood, H. Sussman, S. Manolagas, Characterization of a human osteoblastic osteosarcoma cell line (SAOS-2) with high bone alkaline phosphatase activity. J. Bone Miner. Res. 2(3), 231–238 (1987). https://doi.org/10.1002/jbmr.5650020310. (PMID: 2843003)
X. Wang, E. Tolba, H.C. Schröder, M. Neufurth, Q. Feng, B. Diehl-Seifert, W. Müller, Effect of bioglass on growth and biomineralization of SaOS-2 cells in hydrogel after 3D cell bioprinting. PLoS ONE 9, e112497 (2014). https://doi.org/10.1371/journal.pone.0112497
E. Boccardi, A. Philippart, V. Melli, L. Altomare, L. De Nardo, G. Novajra, A.R. Boccaccini, Bioactivity and mechanical stability of 45S5 bioactive glass scaffolds based on natural marine sponges. Ann. Biomed. Eng. 44(6), 1881–1893 (2016). https://doi.org/10.1007/s10439-016-1595-5
M. Alcaide, P. Portoles, A. Lopez-Noriega, D. Arcos, M. Vallet-Regi, M.T. Portoles, Interaction of an ordered mesoporous bioactive glass with osteoblasts, fibroblasts and lymphocytes, demonstrating its biocompatibility as a potential bone graft material. Acta Biomater. 6(3), 892–899 (2010). https://doi.org/10.1016/j.actbio.2009.09.008
F. Baino, S. Fiorilli, R. Mortera, B. Onida, E. Saino, L. Visai, E. Verne, C. Vitale-Brovarone, Mesoporous bioactive glass as a multifunctional system for bone regeneration and controlled drug release. J. Appl. Biomater. Funct. Mater. 10(1), 12–21 (2012). https://doi.org/10.5301/JABFM.2012.9270
S. Wilkesmann, J. Fellenberg, Q. Nawaz, B.Reible, A. Moghaddam, A.R. Boccaccini, F. Westhauser, Primary osteoblasts, osteoblast precursor cells or osteoblast-like cell lines: which human cell types are (most) suitable for characterizing 45S5-bioactive glass? J. Biomed. Mater. Res. 108A, 663–674 (2020). https://doi.org/10.1002/jbm.a.36846
Acknowledgements
The authors would like to thank The Scientific and Research Council of Turkey (TÜBİTAK) for the financial support provided in the framework of the 1001 program (grant: 219M212). The SEM and XRD analysis were performed at Manisa Celal Bayar University (Turkey)—Applied Science and Research Center (DEFAM).
Funding
Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK). This work was financially supported by The Scientific and Research Council of Turkey (project no: 219M212).
Author information
Authors and Affiliations
Contributions
AMD: conceptualization, methodology, investigation, funding acquisition, writing—original draft, project administration: ME: investigation, formal analysis, visualization: HA: investigation, formal analysis. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deliormanlı, A.M., Ensoylu, M. & Atmaca, H. Preparation of trivalent rare-earth element-substituted bioactive glass robocast scaffolds for osteosarcoma treatment: structural, morphological, mechanical, drug delivery, and biological properties. Appl. Phys. A 130, 174 (2024). https://doi.org/10.1007/s00339-024-07334-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-024-07334-9