
Abstract
Key message
Novel alleles of two reported tiller angle genes and eleven candidate genes for rice tiller angle were identified by combining GWAS with transcriptomic, qRT-PCR and haplotype analysis.
Abstract
Rice tiller angle is a key agronomic trait determining rice grain yield. Several quantitative trait loci (QTLs) affecting rice tiller angle have been mapped in the past decades. Little is known about the genetic base of tiller angle in rice, because rice tiller angle is a complex polygenic trait. In this study, we performed genome-wide association study (GWAS) on tiller angle in rice using a population of 164 japonica varieties derived from the 3 K Rice Genomes Project (3 K RGP). We detected a total of 18 QTLs using 1135519 single-nucleotide polymorphisms (SNP) based on three GWAS models (GLM, FastLMM and FarmCPU). Among them, two identified QTLs, qTA8.3 and qTA8.4, overlapped with PAY1 and TIG1, respectively, and additional 16 QTLs were identified for the first time. Combined with haplotype and expression analyses, we further revealed that PAY1 harbors one non-synonymous variation at its coding region, likely leading to variable tiller angle in the population, and that nature variations in the promoter of TIG1 significantly affect its expression, closely correlating with tiller angle phenotypes observed. Similarly, using qRT-PCR and haplotype analysis, we identified 1 and 7 candidate genes in qTA6.1 and qTA8.1 that were commonly detected by two GWAS models, respectively. In addition, we identified 3 more candidate genes in the remaining 14 novel QTLs after filtering by transcriptome analysis and qRT-PCR. In summary, this study provides new insights into the genetic architecture of rice tiller angle and candidate genes for rice breeding.
Similar content being viewed by others

Introduction
Rice plant architecture is regarded as one of the major agronomical traits that influence grain yield, which is mainly determined by plant height, tiller number, tiller angle and panicle morphology. Tiller angle, mainly controlled by the asymmetric growth of tiller base, is defined as the angle between the main culm and its side tillers, and it is one of the decisive factor for achieving ideal plant architecture in rice (Wang and Li 2008, 2011). In practice, spread-out rice varieties are able to escape from some diseases but occupy too much space. By contrast, extremely compact rice varieties are less efficient in capturing light and are more susceptible to infection by pathogen attack. Thus, a suitable tiller angle is very important for rice yield (Jiang et al. 2012; Huang et al. 2016; Qu et al. 2021).
Previous studies have revealed that rice tiller angle is strongly associated with gravitropism and polar auxin transport (PAT) (Roychoudhry and Kepinski 2015; Zhang et al. 2018; Li et al. 2019). For example, LAZY1 (LA1), the first gene identified in rice, controls tiller angle and encodes a novel plant-specific protein with an unknown function. LA1 controls shoot gravitropism and tiller angle by regulating PAT, affecting the asymmetric distribution of auxin; and the la1-mutant displays enlarged tiller angle in rice (Li et al. 2007). Subsequently, several suppressors of LA1 (SOLs), such as dwarf 17 (d17), d10, d27, d14, and d3, that are involved in the strigolactones (SLs) biosynthetic or signaling pathway are able to recover the enlarged tiller angle phenotype of la1 by enhancing gravitropic response (Sang et al. 2014). Recently, it is reported that LA1-interacting protein OsBRXL4 affects its nuclear localization, and that it is essential for function of LA1 in controlling rice tiller angle. Furthermore, three rice BRXL genes (OsBRXL1, OsBRXL4, and OsBRXL5) can act redundantly in generating the rice tiller angle (Li et al. 2019). In addition, HEAT STRESS TRANSCRIPTION FACTOR 2D (HSFA2D) acts as an upstream positive regulator of LA1-meidated asymmetric distribution of auxin, thus, induces the asymmetric expression of WUSCHEL RELATED HOMEOBOX6 (WOX6) and WOX11, two transcription factors that specify tiller angle by modulating rice shoot gravitropism (Zhang et al. 2018). Other key genes/quantitative trait loci (QTLs) controlling tiller angle have also been identified and functionally characterized over the past decades, including Tiller Angle Control 1 (TAC1) (Yu et al. 2007), PROSTRATE GROWTH 1 (PROG1) (Jin et al. 2008; Tan et al. 2008). Loose Plant Architecture 1 (LPA1) (Wu et al. 2013), PLANT ARCHITECTURE AND YIELD 1 (PAY1) (Zhao et al. 2015), TILLER INCLINED GROWTH 1 (TIG1) (Zhang et al. 2019), TAC3 (Dong et al. 2016a), TAC4 (Li et al. 2021) and LA2 (Huang et al. 2021). Although these findings provide valuable information regarding tiller angle regulation, the underlying molecular mechanisms and functional relationships among them in rice is largely unknown.
Map-based cloning and association mapping have contributed to understanding of the genetic and molecular bases of many complex agronomic traits. However, conventional cloning method is extremely troublesome (Korte and Farlow 2013; Bhat et al. 2021). Fortunately, advances in next-generation sequencing (NGS) technology and bioinformatics tools, providing large-scale SNP arrays in natural groups in rice become reality (Alexandrov et al. 2015; Wang et al. 2020). Genome-wide association studies (GWAS), as a powerful method for studying the genetics of natural variation based on a linkage disequilibrium mapping approach, have widely been applied to detection of complex agronomic traits in plants (Cockram et al. 2010; Wang et al. 2016, 2020; Luján Basile et al. 2019; Okada et al. 2019; Bai et al. 2021). However, so far, only have TAC3 and DWARF2 (D2), been identified as rice tiller angle regulators by GWAS (Dong et al. 2016b). Addition attempts to identify rice tiller angle regulator using GWAS detected several genetic loci (Lu et al. 2015; Wu et al. 2019). These GWAS analyses were performed in indica and japonica varieties or indica varieties (Lu et al. 2015; Dong et al. 2016b; Wu et al. 2019), and few studies have been performed within japonica varieties that generally have smaller tiller angles than indica varieties. To understand the regulatory mechanisms of japonica rice tiller angle, more QTLs need to be identified from the japonica varieties.
The purpose of this study was to detect genetic loci significantly associated with tiller angle in japonica, based on GWAS analysis on a panel of 164 japonica varieties selected from the 3 K RGP, and to explore favorable SNP alleles and reliable candidate genes that can be used to breed rice with ideal tiller angle. Taken together, our study revealed in total 11 candidate genes and provided the basis for further elucidating mechanisms underlying tiller angle in rice.
Materials and methods
Plant materials
The 164 japonica varieties used in this study were derived from were selected from the 3 K RGP (Wang et al. 2018). Detailed information regarding these varieties, including their geographical origin, is shown in Supplementary Table 1.
Genotyping data and SNP filtering
The 3 K RGP 4.8mio SNP dataset was downloaded from the Rice SNP-Seek Database (https://snp-seek.irri.org/) (Alexandrov et al. 2015). A total of 1,135,519 cleaned SNPs were called in the population. SNPs were filtered utilizing the software PLINK (Purcell et al. 2007) with missing rate < 20% and minor allele frequency (MAF) > 0.05.
Evaluation of tiller angle and population structure
All varieties were grown in the experimental field at density of 17 cm × 20 cm. A completely randomized block design with three replicates was performed. Six plants in the middle of every row were selected for evaluation of tiller angle at maturity stage. The mean value of three replicates was used for analysis. The field experiment was performed in December 2015 to April 2016, Sanya city, Hainan (HN) Province, China (18°15′N, 109°30′E). Principal component analysis (PCA) using PLINK (Purcell et al. 2007) was used to assess the population structure.
Genome‑wide association mapping
GWAS was performed on SNPs as described above using the factored spectrally trans- formed linear mixed models (FastLMM) (Lippert et al. 2011), GLM (Wang and Zhang 2021) and FarmCPU (Liu et al. 2016). Suggestive thresholds were calculated using the formula “− log10 (1/effective number of independent SNPs)” as described previously (Wang et al. 2016). The SNP with the minimum P value was considered the lead SNP. Regions were considered as QTLs when SNPs exceeding the P value within a 200-kb interval of the leading SNP. Manhattan plots for the GWAS results were drawn using the R package “qqman” (https://www.r-project.org/).
Haplotype analysis and identification of candidate genes
Candidate genes were scanned within the 200 kb region centered on the lead SNP of each QTL (using the reference Nipponbare genome (http://rice.plantbiology.msu.edu/cgi-bin/gbrowse/rice/). Haplotype analysis of the candidate genes were carried out using non-synonymous SNPs in exons, while the PAY1 and TIG1 were carried out for all available SNPs in the promoter region (2 kb), non-synonymous SNPs in exons, introns, and 3′ untranslated region were selected from the Rice SNP-Seek Database (https://snp-seek.irri.org/). Only haplotypes shared by at least five varieties were used for multiple comparisons. Duncan’s multiple comparison tests followed by a one-way analysis of variance were completed with the R package”agricolae” (https://myaseen208.github.io/agricolae/https://cran.r-project.org/package=agricolae).
Quantitative reverse transcription PCR (qRT‑PCR) analysis
Tiller base from the varieties used in this study, were collected in triplicate at the tillering stage.
Total RNA was extracted using the Trizol reagent (Invitrogen), following the manufacturer’s instructions. cDNA was synthesized from total RNA using the Fast-Quant RT Kit (with gDNase; Tiangen). Quantitative real time PCR (qRT-PCR) was performed in a two-step reaction using SuperReal PreMix Color (SYBR Green; Tiangen) on a Roche Light Cycler 2.10 system with three technical replicates. Expression levels were normalized to OsActin1 (LOC_Os03g50885).
The sequences of the candidate genes were downloaded from the Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/analyses_search_locus.shtml). Primer sequences for candidate genes were downloaded from the qPrimerDB-qPCR Primer Database (https://biodb.swu.edu.cn/qprimerdb/best-primers-ss) and designed by NCBI except for LOC_Os08g05900, which could not find the appropriate primer. Primer sequences are given in Supplementary Table 6.
Results
Natural variation of rice tiller angle
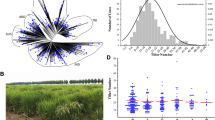
To dissect the genetic structure of rice tiller angle, a total of 164 japonica varieties from 3 K RGP were selected for tiller angle phenotyping at the heading stage in Hainan, China (Supplementary Table 1). The tiller angle observed at the maturity stage in this panel ranged from 6.3° to 35.6°, which exhibited a nearly normal distribution (Fig. 1a), indicating that tiller angle was regulated by many genes with small or moderate effects (Zhang et al. 2018; He et al. 2021). Tiller angles of about half of the panel (46.2%, 73 of 164) were between 18 and 24˚, with the median value of 19.4˚ (Fig. 1a, b). Principal component analysis (PCA) of the 164 varieties using high-confidence SNPs showed that all tested varieties are closely linked, and that there is no obvious structural pattern existed in this panel (Supplementary Fig. 1), avoiding population structure-induced false positives. Taken together, these results indicate that the rice panel used in this study has abundant genetic diversity and is suitable for GWAS.

Tiller angle of the 164 japonica varieties. a Distributions of tiller angle. b Box plot of the phenotypic variation of tiller angle. Boxes show median, and upper and lower quartiles. Whiskers extend to 1.5 × the interquartile range, with any remaining points indicated with dots. n number of varieties
GWAS for tiller angle
To dissect all possible genomic loci associated with tiller angle, we performed GWAS with FastLMM, FarmCPU and GLM models using 1135519 high-confidence SNPs. The genome-wide suggestive thresholds P ≤ 8.81E− 7 were calculated using the formula “− log10 (1/effective number of independent SNPs)”. Totally, 18 QTLs were identified to be significantly associated with tiller angle when significant SNPs located within 200-kb range (Fig. 2). Among these QTLs, 8, 3, 9 QTLs were identified by GLM (Wang and Zhang 2021), FastLMM (Lippert et al. 2011) and FarmCPU (Liu et al. 2016), respectively. Notably, 2 QTLs, qTA8.3 and qTA8.4, identified in this study were co-localized with PAY1 (Zhao et al. 2015) and TIG1 (Zhang et al. 2019), respectively (Table 1), confirmed the reliability of our GWAS results. Among the remaining 16 QTLs newly identified, 2 QTLs (qTA6.1 and qTA8.1) were simultaneously identified by both GLM and FastLMM approaches, which are highly valuable for further identification of the causal genes for tiller angle trait.

Manhattan plots of GWAS for tiller angle in 164 rice varieties. The arrows represent QTLs (red arrows for single GWAS model detected QTLs, green arrows for multiple GWAS models detected QTLs and black arrows for reported genes) when the peak SNP correlation exceeds the significance threshold, − log10(P) > 6.06 (P < (P ≤ 8.81E − 7)
PAY1, the candidate gene of qTA8.3
The QTL qTA8.3 was found to contain PAY1 gene, a single nucleotide change, G to A, at position 19468238 in exon 4 of which affected plant architecture via affecting polar auxin transport activity and altering endogenous indole-3-acetic acid distribution (Zhao et al. 2015). To understand how PAY1 sequence may affect tiller angle phenotype, we analyzed SNPs in the genomic coding region and 2 kb of upstream promoter region of PAY1 across 164 varieties, which revealed 2 major haplotypes; tiller angles of varieties containing PAY1HapA were significantly lower than those of PAY1 HapB, four SNP variations including only one in the first exon were observed between these two haplotypes (Fig. 3a, b; Supplementary Table 1). However, none of those variations observed in these two haplotypes was the same as the functional mutation previously reported in PAY1 (Zhao et al. 2015). The SNP variations observed in both promoter region and CDS region of PAY1 indicated that both may play important role in regulating tiller angle in rice. To further investigate the causative SNP variation in PAY1 responsible for tiller angle, we used qRT-PCR to examine expression level of PAY1 between varieties harboring PAY1HapA or PAY1HapB, and results showed that expression levels of PAY1 are not associated with haplotypes (Fig. 3c), indicating that the only variation in the first exon, a non-synonymous SNP (T to C) identified in PAY1 could be a new functional allele that leads to spread phenotypes. In general, japonica varieties always have a relatively smaller tiller angle and show a more compact plant architecture than indica varieties (Dong et al. 2016a). Therefore, to further understand the breeding utilization of “T” and “C” alleles in PAY1 between the japonica and indica varieties. we investigated the changes of allelic frequencies using 2584 varieties from 3 K RGP (Fig. 3d; Supplementary Table 2). The results showed that nearly all of the indica varieties (98%) contained “C” allele, while most japonica varieties (73%) contained “T” allele, indicating that PAY1 has been selected during japonica-indica differentiation.

Haplotype analysis of PAY1. a Two haplotypes of PAY1 based on 4 SNPs observed in all assessed rice varieties. The schematic representation of PAY1 gene structure (upper) shows the promoter as a white box, exons as green boxes, and introns and intergenic regions as black lines. Thin black lines indicate the genomic position of each SNP. Haplotypes with fewer than 5 varieties are not shown. The SNP in red and bold is a non- synonymous SNP. ‘$#’ indicates a missense mutation from Trp to Arg. b, c Box plots for b tiller angle based on the two haplotypes for PAY1 and c PAY1 expression levels in different haplotypes relative to OsActin1. Boxes show median, and upper and lower quartiles. Whiskers extend to 1.5 × the interquartile range, with any remaining points indicated with dots. *P < 0.05 (Welch two sample t test). d Frequencies of the two different alleles in PAY1 between the japonica and indica varieties. n, number of varieties in each panel allele
TIG1, the candidate gene of qTA8.4
Another QTL, qTA8.4, contained TIG1, which encodes a TCP family transcriptional activator and variations in the TIG1 promoter of indica varieties (tig1 allele) decrease expression of TIG1 in the adaxial side of tiller base and reduce cell length and tiller angle, leading to the transition from inclined tiller growth in wild rice to erect tiller growth during rice domestication (Zhang et al. 2019). To investigate the causative SNP variations in TIG1 responsible for phenotypic variations for tiller angle, we analyzed SNPs in the genomic coding region and 2 kb of upstream promoter region of TIG1 across 164 varieties, which revealed 4 major haplotypes with no SNP in CDS region (Fig. 4a; Supplementary Table 1). Compared with previously reported SNP variations of TIG1 in rice (Zhang et al. 2019), SNPs detected in our research were completely new. Moreover, multiple comparison tests of tiller angle showed that tiller angles of varieties with TIG1HapA were significantly higher than those with TIG1HapC or TIG1HapD (Fig. 4b), indicating that SNP variations identified in promoter regions may play an important role in regulating tiller angle as a new allele. To test whether the relative expression level of different haplotypes of TIG1 were associated with tiller angle, we used qRT-PCR analysis on samples from tiller base, which revealed that expression level of TIG1 in TIG1HapA varieties was significantly higher than that in TIG1HapD varieties (Fig. 4c), in agreement with previously report that TIG1 positively regulates tiller angle in rice (Zhang et al. 2019).

Haplotype analysis of TIG1. a Four haplotypes of TIG1 based on 12 SNPs observed in all assessed rice varieties. The schematic representation of TIG1 gene structure (upper) shows the promoter as a white box, exons as green boxes, and intergenic regions as black lines. Thin black lines indicate the genomic position of each SNP. Haplotypes with fewer than 5 varieties are not shown. Yellow highlight indicates SNP alternatives. b, c Box plots for b tiller angle based on the four haplotypes for TIG1 and c TIG1 expression levels in different haplotypes relative to OsActin1. Boxes show median, and upper and lower quartiles. Whiskers extend to 1.5 × the interquartile range, with any remaining points indicated with dots. **P < 0.01 (Welch two sample t test). Letters indicate significant differences, P < 0.05 (Duncan’s multiple comparison test)
Candidate genes analysis of qTA6.1 and qTA8.1 by qRT-PCR and haplotype analysis
To identify the candidate genes in two novel QTLs (qTA6.1 and qTA8.1) that were commonly detected by both GLM and FastLMM, we selected candidate genes in a 200-kb region (100 kb flanking the left and right side of the most significantly associated SNPs) using the Nipponbare reference genome (http://rice.plantbiology.msu.edu), and found contained 60 genes including 50 functionally annotated genes (31, 19 in qTA6.1 and qTA8,1, respectively) and 10 retro/transposon/hypothetical proteins (Supplementary Table 3). Two analyses were performed further to identify candidate genes for these two QTLs: (1) expression analysis using qRT-PCR analysis in the tiller base of 22 varieties: 11 spread-tiller varieties and 11 compact-tiller varieties; (2) haplotype analyses in CDS region.
For qTA6.1, LOC_Os06g06990 was the only one showing significantly lower expression level in spread-tiller varieties than compact-tiller varieties as revealed by qRT-PCR analysis (Fig. 5a). Haplotype analysis in CDS region of all candidate genes in the same region showed that 12 genes can be divided into different haplotypes, and that none of identified SNP variations among these genes were associated with significant differences in the tiller angle (Supplementary Fig. 2), indicating that the LOC_Os06g06990 is likely the most promising candidate gene for qTA6.1.


The candidate genes analysis in qTA6.1 and qTA8.1. a, b The relative expression of LOC_Os06g06990 and LOC_Os08g06090 in extreme tiller angle phenotypes relative to OsActin1. The green horizontal lines depict the average expression levels in the spread-tiller varieties and compact-tiller varieties, respectively. c Haplotype analyses of the candidate genes Gene structures (upper) and different haplotypes (lower). Thin black lines indicate the genomic position of each SNP. Haplotypes with fewer than 5 varieties are not shown. Boxes show median, and upper and lower quartiles. Whiskers extend to 1.5 × the interquartile range, with any remaining points indicated with dots. *P < 0.05, ***P < 0.001 (Welch two sample t test)
For qTA8.1, LOC_Os08g06090 was shown to be significantly and highly expressed in spread-tiller varieties than that in compact-tiller varieties as evidenced by qRT-PCR analysis (Fig. 5b). Additional haplotype analysis in CDS region showed that 6 genes (LOC_Os08g05840, LOC_Os08g05860, LOC_Os08g05870, LOC_Os08g05890, LOC_Os08g05900, and LOC_Os08g05910) can be divided into different haplotypes, all were associated with significant differences in the tiller angle (Fig. 5c). These results indicated that abovementioned seven genes are all potential candidate genes for qTA8.1.
Candidate genes analysis of the remaining 14 novel QTLs by integrating GWAS and transcriptomics
We further analyzed the 100 kb window near the leading SNPs within these remaining 14 QTLs. After removing 142 retro/transposon/hypothetical proteins, 297 annotated genes were identified (Supplementary Table 4).
To further narrow down candidate gene numbers, we referred to two publicly available transcriptomic data about tiller angle regulatory genes: one contained 4204 differentially expressed genes (DGEs) between gravistimulated shoots and control shoots without stimulation (Zhang et al. 2018); the other contained 4431 DGEs between ZnF transgenic plants and the wild type (Wu et al. 2018). By combining these DEGs and our GWAS results, we finally identified 15 overlapping candidate genes (Fig. 6a). Further qRT-PCR analysis showed that among these 15 candidate genes, expression levels of 3 genes (LOC_Os01g21590, LOC_Os03g15050 and LOC_Os04g41229) are significantly lower in spread-tiller varieties than in compact-tiller varieties (Fig. 6b and Supplementary Table 5), showing close association with tiller angle. These 3 candidate genes are likely to be a promising candidate gene in qTA1.2, qTA3.1 and qTA4.1, respectively, negatively regulating the tiller angle in rice.


Candidate gene identification by integrating GWAS and transcriptomic analysis. a Venn diagram showed that a total 15 overlapping candidate genes were identified. b The relative expression of LOC_Os01g21590, LOC_Os03g15050 and LOC_Os04g41229 in extreme tiller angle phenotypes relative to OsActin1. The green horizontal lines depict the average expression levels in the spread-tiller varieties and compact-tiller varieties, respectively. *P < 0.05 (Welch two sample t test)
Discussion
Natural variation, including phenotypic and genotypic variation caused by spontaneous mutations and sections, has been maintained in nature by all evolutionary processes (Jiang et al. 2012). Rice tiller angle is controlled by QTLs derived from natural variations (Yu et al. 2007; Jin et al. 2008; Tan et al. 2008; Jiang et al. 2012; Dong et al. 2016a; Zhang et al. 2019). Therefore, dissection of new genetic variation controlling tiller angle is important for rice breeding to use potential source of beneficial alleles. Compared with QTL mapping, GWAS enables the identification of causal loci at high-resolution and is a powerful tool for detecting favorable alleles in natural resources (Ogura and Busch 2015; Bai et al. 2021). In this study, 164 japonica varieties were selected from 3 K RGP for studying the genetic basis of tiller angle using GWAS as well as other approaches. Our findings provide important novel genetic resource for further gene discovery and breeding of tiller angle in rice.
Phenotype variations and GWAS
The erect plant architecture is favored by human beings, because it improves photosynthetic efficiency and increases planting density, which eventually enhances grain yield (Wang and Li 2008, 2011). The genetic basis of rice tiller angle is complex and is associated with many loci, each with small contributions to phenotypic variance (Lu et al. 2015; Wu et al. 2019), our results confirmed these previous findings and showed rice tiller angle trait tends to show a nearly normal distribution in a segregated population (Fig. 1a). The natural variation of rice tiller angle revealed in this study, ranging from 6.3° to 35.6°(Fig. 1a, b), indicated not only that the panel selected in this study is rich in genetic diversity that suitable for GWAS analysis, but also that this panel is useful for direct use as parents in rice breeding for ideal tiller angle.
We used three models to perform GWAS in this study, all generated QTL signals, some of them were repeatedly detected in more than one model, while most of them were not (Fig. 2). These results indicated that when performing GWAS, if possible, different models should be used to identify as much as possible the QTLs. Our results are consistent with previous report that different GWAS analysis methods possess unique features and complement each other (Liu et al. 2020). In total, three models yielded 18 QTLs for tiller angle; and the identification of two known rice tiller angle genes, PAY1 and TIG1, that co-localized with qTA8.3 and qTA8.4, respectively, indicated the reliability of our GWAS results.
Novel alleles in PAY1 and TIG1 affect tiller angle in rice
Some alleles of the genes related to yield-related QTLs were selected during domestication or breeding programs (Jin et al. 2008; Tan et al. 2008; Jiang et al. 2012). The allele mining technique has been successfully employed to find important variations at various locus for understanding mechanisms underlying domestication and for breeding (Huang et al. 2018; Guo et al. 2020; Bai et al. 2021). Present study identified novel alleles of PAY1 and TIG1 through GWAS in a japonica panel that are associated with variable tiller angle, and provided genetic resource for further understanding mechanisms underlying rice tiller angle. Clearly, the two alleles have different mechanisms regulating rice tiller angle. The SNP in coding region of PAY1 plays important role in regulating tiller angle (Fig. 3), while SNPs in the promoter region of TIG1 are associated with variation of tiller angle (Fig. 4). Therefore, learning from domestication, the promoter edited are interesting targets for genome editing to create new alleles for rice breeding as did previously (Swinnen et al. 2016; Oliva et al. 2019; Huang et al. 2020). Moreover, differences in allelic frequencies in the two sub-species (Fig. 3d) suggest that PAY1 has been subjected to selection during rice breeding, which partially explains why the Indica varieties usually have a relatively larger tiller angle and display comparatively spread-out plant architecture, whereas japonica varieties always have a relatively smaller tiller angle and show a more compact plant architecture (Yu et al. 2007; Zhang et al. 2019). Similarly, the major tiller angle gene TIG1, is reported to be strongly selected in indica varieties by human beings (Zhang et al. 2019). Therefore, our study suggests the idea that rice tiller angle is selected during domestication.
Notably, the previous reported variants in both PAY1 and TIG1 were not found in 3 K RGP (Zhao et al. 2015; Zhang et al. 2019), the detected SNPs, particularly those in the TIG1 promoter, in this study, thus, provide valuable information for breeding rice varieties, especially japonica rice.
Novel candidate genes
Previous studies have demonstrated that rice tiller angle is strongly related to plant gravitropic responses, which also can be effected by hormones, such as auxin and brassinosteroids (BRs) (Dong et al. 2016a; Liu et al. 2016; Waite and Dardick 2018; Zhang et al. 2018). Using expression and haplotype analysis combined with GWAS, we identified 11 candidate genes for rice tiller angle in this study (Fig. 5a, c and Fig. 6b). Among them, 4 candidate genes (LOC_Os08g05840, LOC_Os08g05870, LOC_Os08g05890 and LOC_Os01g21590) attracted specific attention based on their functional annotations. Because DNA Topoisomerase 1 (OsTOP1) functions as a role in gravitropism by linking transcriptional R-loops with auxin signaling (Shafiq et al. 2017), LOC_Os08g05840, encoding DNA topoisomerase 1, likely is involved in tiller angle regulation in rice. LOC_Os08g05840 may be regulation of tiller angle in rice. LOC_Os08g05870 and LOC_Os08g05890 encode an oxysterol-binding protein-related protein 6 and a MSP domain containing protein, respectively. both protein families have been documented to transport sterols and involved in sterol regulation (Reynolds and Hand 2009; Saravanan et al. 2009; Umate 2011), Considering that sterols also act as precursors for BR hormones, we propose that LOC_Os08g05870 and LOC_Os08g05890 are two important components of BR signaling associated with rice tiller angle. LOC_Os01g21590, encodes a homeodomain containing protein, whose homologues have been reported to be associated with tiller angle (Zhang et al. 2018; Hu et al. 2020). Considering its significant higher expression in compact-tiller varieties than in spread-tiller varieties (Fig. 6b), LOC_Os01g21590 is likely connected with tiller angle in rice. Nevertheless, these candidate genes provide useful genetic resources for expand our understanding regarding hormone mediated tiller angle regulation in rice.
In summary, this study provides important genetic information for better understanding the genetic diversity of tiller angle in rice and useful genetic resources for breeding rice varieties with ideal plant architecture.
Availability of data and materials
The association mapping population data can be found in Rice SNP-Seek database (https://snp-seek.irri.org/), and transcriptome data can be found in the previously studies supplementary data (Wu et al. 2018; Zhang et al. 2018).
Code availability
Not applicable.
References
Alexandrov N, Tai S, Wang W et al (2015) SNP-seek database of SNPs derived from 3000 rice genomes. Nucleic Acids Res 43:D1023–D1027. https://doi.org/10.1093/nar/gku1039
Bai S, Hong J, Li L et al (2021) Dissection of the genetic basis of rice panicle architecture using a genome-wide association study. Rice. https://doi.org/10.1186/s12284-021-00520-w
Bhat JA, Yu D, Bohra A et al (2021) Features and applications of haplotypes in crop breeding. Commun Biol 4:1–12. https://doi.org/10.1038/s42003-021-02782-y
Cockram J, White J, Zuluaga DL et al (2010) Genome-wide association mapping to candidate polymorphism resolution in the unsequenced barley genome. Proc Natl Acad Sci U S A 107:21611–21616. https://doi.org/10.1073/pnas.1010179107
Dong H, Zhao H, Xie W et al (2016a) A novel tiller angle gene, TAC3, together with TAC1 and D2 largely determine the natural variation of tiller angle in rice cultivars. PLoS Genet 12:1–21. https://doi.org/10.1371/journal.pgen.1006412
Dong H, Zhao H, Xie W et al (2016b) A novel tiller angle gene, TAC3, together with TAC1 and D2 largely determine the natural variation of tiller angle in rice cultivars. PLoS Genet. https://doi.org/10.1371/journal.pgen.1006412
Guo H, Zeng Y, Li J et al (2020) Differentiation, evolution and utilization of natural alleles for cold adaptability at the reproductive stage in rice. Plant Biotechnol J 18:2491–2503. https://doi.org/10.1111/pbi.13424
He Y, Li L, Jiang D (2021) Understanding the regulatory mechanisms of rice tiller angle, then and now. Plant Mol Biol Rep 39:640–647. https://doi.org/10.1007/s11105-021-01279-6
Hu Y, Li S, Fan X et al (2020) OsHOX1 and OsHOX28 redundantly shape rice tiller angle by reducing HSFA2D expression and auxin content. Plant Physiol 184:1424–1437. https://doi.org/10.1104/pp.20.00536
Huang L, Li Q, Zhang C et al (2020) Creating novel Wx alleles with fine-tuned amylose levels and improved grain quality in rice by promoter editing using CRISPR/Cas9 system. Plant Biotechnol J 18:2164–2166. https://doi.org/10.1111/pbi.13391
Huang L, Wang W, Zhang N et al (2021) LAZY2 controls rice tiller angle through regulating starch biosynthesis in gravity-sensing cells. New Phytol 231:1073–1087. https://doi.org/10.1111/nph.17426
Huang X, Yang S, Gong J et al (2016) Genomic architecture of heterosis for yield traits in rice. Nature 537:629–633. https://doi.org/10.1038/nature19760
Huang Y, Zhao S, Fu Y et al (2018) Variation in the regulatory region of FZP causes increases in secondary inflorescence branching and grain yield in rice domestication. Plant J 96:716–733. https://doi.org/10.1111/tpj.14062
Jiang J, Tan L, Zhu Z et al (2012) Molecular evolution of the TAC1 gene from rice (Oryza sativa L.). J Genet Genom 39:551–560. https://doi.org/10.1016/j.jgg.2012.07.011
Jin J, Huang W, Gao JP et al (2008) Genetic control of rice plant architecture under domestication. Nat Genet 40:1365–1369. https://doi.org/10.1038/ng.247
Korte A, Farlow A (2013) The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9:1–9. https://doi.org/10.1186/1746-4811-9-29
Li Z, Liang Y, Yuan Y et al (2019) OsBRXL4 regulates shoot gravitropism and rice tiller angle through affecting LAZY1 nuclear localization. Mol Plant 12:1143–1156. https://doi.org/10.1016/j.molp.2019.05.014
Li H, Sun H, Jiang J et al (2021) TAC4 controls tiller angle by regulating the endogenous auxin content and distribution in rice. Plant Biotechnol J 19:64–73. https://doi.org/10.1111/pbi.13440
Li P, Wang Y, Qian Q et al (2007) LAZY1 controls rice shoot gravitropism through regulating polar auxin transport. Cell Res 17:402–410. https://doi.org/10.1038/cr.2007.38
Lippert C, Listgarten J, Liu Y et al (2011) FaST linear mixed models for genome-wide association studies. Nat Methods 8:833–835. https://doi.org/10.1038/nmeth.1681
Liu JM, Park SJ, Huang J et al (2016) Loose plant architecture1 (LPA1) determines lamina joint bending by suppressing auxin signalling that interacts with C-22-hydroxylated and 6-deoxo brassinosteroids in rice. J Exp Bot 67:1883–1895. https://doi.org/10.1093/jxb/erw002
Liu S, Zhong H, Meng X et al (2020) Genome-wide association studies of ionomic and agronomic traits in USDA mini core collection of rice and comparative analyses of different mapping methods. BMC Plant Biol 20:441. https://doi.org/10.1186/s12870-020-02603-0
Lu Q, Zhang M, Niu X et al (2015) Genetic variation and association mapping for 12 agronomic traits in indica rice. BMC Genomics 16:1–17. https://doi.org/10.1186/s12864-015-2245-2
Luján Basile SM, Ramírez IA, Crescente JM et al (2019) Haplotype block analysis of an argentinean hexaploid wheat collection and GWAS for yield components and adaptation. BMC Plant Biol 19:1–16. https://doi.org/10.1186/s12870-019-2015-4
Ogura T, Busch W (2015) From phenotypes to causal sequences: using genome wide association studies to dissect the sequence basis for variation of plant development. Curr Opin Plant Biol 23:98–108. https://doi.org/10.1016/j.pbi.2014.11.008
Okada Y, Monden Y, Nokihara K et al (2019) Genome-wide association studies (GWAS) for yield and weevil resistance in sweet potato (Ipomoea batatas (L.) Lam). Plant Cell Rep 38:1383–1392. https://doi.org/10.1007/s00299-019-02445-7
Oliva R, Ji C, Atienza-Grande G et al (2019) Broad-spectrum resistance to bacterial blight in rice using genome editing. Nat Biotechnol 37:1344–1350. https://doi.org/10.1038/s41587-019-0267-z
Purcell S, Neale B, Todd-Brown K et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575. https://doi.org/10.1086/519795
Qu M, Zhang Z, Liang T et al (2021) Overexpression of a methyl-CpG-binding protein gene OsMBD707 leads to larger tiller angles and reduced photoperiod sensitivity in rice. BMC Plant Biol 21:1–14. https://doi.org/10.1186/s12870-021-02880-3
Reynolds JA, Hand SC (2009) Embryonic diapause highlighted by differential expression of mRNAs for ecdysteroidogenesis, transcription and lipid sparing in the cricket Allonemobius socius. J Exp Biol 212:2075–2084. https://doi.org/10.1242/jeb.027367
Roychoudhry S, Kepinski S (2015) Shoot and root branch growth angle control-the wonderfulness of lateralness. Curr Opin Plant Biol 23:124–131. https://doi.org/10.1016/j.pbi.2014.12.004
Sang D, Chen D, Liu G et al (2014) Strigolactones regulate rice tiller angle by attenuating shoot gravitropism through inhibiting auxin biosynthesis. Proc Natl Acad Sci U S A 111:11199–11204. https://doi.org/10.1073/pnas.1411859111
Saravanan RS, Slabaugh E, Singh VR et al (2009) The targeting of the oxysterol-binding protein ORP3a to the endoplasmic reticulum relies on the plant VAP33 homolog PVA12. Plant J 58:817–830. https://doi.org/10.1111/j.1365-313X.2009.03815.x
Shafiq S, Chen C, Yang J et al (2017) DNA topoisomerase 1 prevents r-loop accumulation to modulate auxin-regulated root development in rice. Mol Plant 10:821–833. https://doi.org/10.1016/j.molp.2017.04.001
Swinnen G, Goossens A, Pauwels L (2016) Lessons from domestication: targeting cis-regulatory elements for crop improvement. Trends Plant Sci 21:506–515. https://doi.org/10.1016/j.tplants.2016.01.014
Tan L, Li X, Liu F et al (2008) Control of a key transition from prostrate to erect growth in rice domestication. Nat Genet 40:1360–1364. https://doi.org/10.1038/ng.197
Umate P (2011) Oxysterol binding proteins (OSBPs) and their encoding genes in Arabidopsis and rice. Steroids 76:524–529. https://doi.org/10.1016/j.steroids.2011.01.007
Waite JM, Dardick C (2018) Tiller angle control 1 modulates plant architecture in response to photosynthetic signals. J Exp Bot 69:4935–4944. https://doi.org/10.1093/jxb/ery253
Wang Y, Li J (2008) Molecular basis of plant architecture. Annu Rev Plant Biol 59:253–279. https://doi.org/10.1146/annurev.arplant.59.032607.092902
Wang Y, Li J (2011) Branching in rice. Curr Opin Plant Biol 14:94–99. https://doi.org/10.1016/j.pbi.2010.11.002
Wang W, Mauleon R, Hu Z et al (2018) Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 557:43–49. https://doi.org/10.1038/s41586-018-0063-9
Wang X, Wang H, Liu S et al (2016) Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat Genet 48:1233–1241. https://doi.org/10.1038/ng.3636
Wang Q, Tang J, Han B, Huang X (2020) Advances in genome-wide association studies of complex traits in rice. Theor Appl Genet 133:1415–1425. https://doi.org/10.1007/s00122-019-03473-3
Wang J, Zhang Z (2021) GAPIT version 3: boosting power and accuracy for genomic association and prediction. Genom Proteom Bioinform. https://doi.org/10.1016/j.gpb.2021.08.005
Wu D, Guo Z, Ye J et al (2019) Combining high-throughput micro-CT-RGB phenotyping and genome-wide association study to dissect the genetic architecture of tiller growth in rice. J Exp Bot 70:545–561. https://doi.org/10.1093/jxb/ery373
Wu X, Tang D, Li M et al (2013) Loose plant architecture1, an indeterminate domain protein involved in shoot gravitropism, regulates plant architecture in rice. Plant Physiol 161:317–329. https://doi.org/10.1104/pp.112.208496
Wu Y, Zhao S, Li X et al (2018) Deletions linked to PROG1 gene participate in plant architecture domestication in Asian and African rice. Nat Commun 9:1–10. https://doi.org/10.1038/s41467-018-06509-2
Yu B, Lin Z, Li H et al (2007) TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J 52:891–898. https://doi.org/10.1111/j.1365-313X.2007.03284.x
Zhang W, Tan L, Sun H et al (2019) Natural variations at TIG1 encoding a TCP transcription factor contribute to plant architecture domestication in rice. Mol Plant 12:1075–1089. https://doi.org/10.1016/j.molp.2019.04.005
Zhang N, Yu H, Yu H et al (2018) A core regulatory pathway controlling rice tiller angle mediated by the LAZY1-dependent asymmetric distribution of auxin. Plant Cell 30:1461–1475. https://doi.org/10.1105/tpc.18.00063
Zhao L, Tan L, Zhu Z et al (2015) PAY1 improves plant architecture and enhances grain yield in rice. Plant J 83:528–536. https://doi.org/10.1111/tpj.12905
Acknowledgements
We would like to thank Mr. Zhijing Luo and Ms. Mingjiao Chen for their assistance in rice growth.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
DZ and WL designed the study. SB and JH performed the experiments and bioinformatic analysis. ZL and WW contributed plant materials. JS helped to revise the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Additional information
Communicated by Zheng-Yi Xu.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bai, S., Hong, J., Su, S. et al. Genetic basis underlying tiller angle in rice (Oryza sativa L.) by genome-wide association study. Plant Cell Rep 41, 1707–1720 (2022). https://doi.org/10.1007/s00299-022-02873-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-022-02873-y