
Abstract
Background
This phase 1 study evaluated the safety, tolerability, pharmacokinetics and efficacy of patritumab (U3-1287) Process 2, a new formulation of fully human anti-HER3 monoclonal antibody in combination with erlotinib, an epidermal growth factor receptortyrosine kinase inhibitor (EGFR-TKI) in prior chemotherapy treated Japanese patients with advanced non-small cell lung cancer (NSCLC).
Methods
Patients received intravenous patritumab Process 2 formulation at 9 mg/kg every 3 weeks after initiation of 18 mg/kg loading dose combined with continuous daily dose of erlotinib (150 mg QD) until any of the withdrawal criteria are met. Adverse events (AEs) were assessed using CTCAE v4.0 and tumor response was assessed using RECIST v1.1. Full pharmacokinetic sampling and serum biomarker analyses were mainly performed during cycle 1 and 2.
Results
Total of six EGFR-mutant NSCLC patients including one EGFR-TKI naïve patient received patritumab Process 2 formulation combined with erlotinib. No dose-limiting toxicities were observed. The most frequent AEs were gastrointestinal or skin toxicities, which were generally mild and manageable. One patient discontinued from study due to reversible grade 3 interstitial lung disease. The mean area under the curve (AUC) value was 2640 μg/day/mL; the Cmax value was 434 μg/mL, respectively. The median progression-free survival (95% confidence interval) was 220.0 (100.0–363.0) days. HER3 ligand heregulin was detected in serum from only a patient that maintained most durable stable disease.
Conclusions
Patritumab Process 2 formulation in combination with erlotinib was well tolerated compatible with favorable PK profile in Japanese patients with advanced NSCLC.
Similar content being viewed by others

Introduction
The human epidermal growth factor receptor-3 (HER3) is a member of the HER (EGFR/ErbB) receptor family consisting of four closely related type 1 transmembrane receptors (EGFR, HER2, HER3, and HER4). HER receptors are part of a complex signaling network intertwined with the Ras/Raf/MAPK, PI3K/AKT, JAK/STAT, and PKC signaling pathways. HER3 is expressed in many normal tissues and in a variety of solid tumors, including non-small cell lung cancer (NSCLC) [1–4], and increased levels of HER3 have been associated with a negative clinical prognosis, including survival in several tumor types [5–8]. HER3 is the only HER family member that lacks tyrosine kinase activity because of an amino acid substitution in the conserved kinase domain. Thus, interactions of HER3 with binding partners are essential for its biological activity [9]. In particular, HER3 potently activates downstream phosphatidylinositol-3-kinase (PI3K) and AKT pathway signaling by directly binding to PI3K through six consensus phosphotyrosinesites [10]. Recent preclinical and clinical data also suggest that HER3 is involved in resistance to other HER receptor-targeted therapeutics [11–15]. Since HER3 has limited kinase activity, several newly developed monoclonal antibodies (mAbs) are being explored to target HER3 for cancer therapy.
Patritumab (U3-1287) is a fully human monoclonal immunoglobulin G1 (IgG1) antibody directed against HER3, thereby inhibiting ligand binding [heregulin alpha (HRG-α) and heregulin beta (HRG-β)] and receptor activation and induces HER3 down-regulation. Functionally, patritumab inhibits tumor cell proliferation, survival, and anchorage-independent growth in vitro, and inhibits growth of HER3 expressing xenograft tumor models in vivo [16–19]. In addition, the combined use of patritumab with erlotinib, an epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI), led to increased inhibition of tumor proliferation, compared with patritumab alone [18].
In a first-in-human phase 1 study (ClinicalTrials.gov Identifier: NCT00730470), the safety and tolerability of patritumab were evaluated up to the dose of 20 mg/kg without dose-limiting toxicities (DLTs) [20]. In another phase 1 study (ClinicalTrials.jp Identifier: JapicCTI-101,262) in Japanese patients, patritumab was well tolerated up to 18 mg/kg without DLTs and PK profile was similar to the US phase I study [21]. Regarding the combined approaches of former formulation of patritumab (patritumab Process 1) with other molecular targeting agents, both erlotinib in patients with NSCLC and trastuzumab and paclitaxel in patients with HER2 overexpressing metastatic breast cancer were evaluated [22, 23]. The drug substance and product for use in nonclinical studies, phase 1 clinical studies, and phase 2 clinical studies during early phases of development of patritumab were manufactured using Manufacturing Method Process 1. Subsequently, a new method, Process 2 was developed to manufacture the drug substance and product for use in a global phase 3 study, with the aim of increasing the yield of the target protein and improving properties of the drug substance and/or product. Based on both safety profile and clinical activities of patritumab Process 1 combined with EGFR-TKI, erlotinib in patients with advanced NSCLC, this study evaluated that the safety and pharmacokinetics of patritumab Process 2 in combination with erlotinib and potential biomarkers related to patritumab were also evaluated.
Materials and methods
Patient eligibility
This study was conducted based on the Declaration of Helsinki and the Guidelines for the Clinical Evaluation Methods of Anti-Cancer Drugs in Japan (Japanese Ministry of Health, Labour, and Welfare notification, November 1, 2005). The study was approved by the institutional review board of study site.
The main eligibility criteria were as follows: a histologically or cytologically confirmed diagnosis of stage IIIB/IV NSCLC in a patient who had experienced disease progression while on the standard therapy or in a patient intolerant of, or not eligible for the standard therapy (prior EGFR-TKIs therapy allowed); a patient age ≥ 20 years; an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1; a life expectancy of more than 3 months; and adequate hematologic, hepatic, and renal functions. The exclusion criteria included the administration of chemotherapy, radiotherapy, or biological therapy in the 4 weeks (2 weeks for palliative radiotherapy and kinase inhibitors) prior to enrollment; other active malignancies; history or presence of interstitial lung disease (ILD); history (within 6 months before enrollment) or presence of severe cardiovascular or cerebrovascular disease, pulmonary thrombosis, deep vein thrombosis, or other clinically severe pulmonary disease; any of the following complications, including clinically severe infections requiring systemic administration of an antimicrobial agent, antiviral agent or other agents; presence of chronic diarrhea, inflammatory bowel disease or partial ileus; presence of peptic ulcer; fluid retention requiring treatment; corneal disease; uncontrolled diabetes mellitus; hypertension; psychiatric symptoms; appositive test for hepatitis B virus surface antigen, hepatitis C virus antibody or human immunodeficiency virus antibody; history of a bleeding diathesis; and history of serious hypersensitivity to drugs containing polysorbate 20. All patients provided informed consent, and the study was conducted in accordance with the current Good Clinical Practice standards. This study was registered at ClinicalTrials.jp Identifier:JapicCTI-152,841.
Study design and evaluation
This study was an open-label, non-randomized, phase 1 study of patritumab Process 2 formulation of fully human anti-HER3 monoclonal antibody in combination with erlotinib in Japanese patients with advanced NSCLC that was conducted at single site in Japan. The primary objective was to evaluate the safety and tolerability of patritumab Process 2 formulation combined with erlotinib in Japanese patients with advanced NSCLC. Secondary objectives were to assess the PK profile, preliminary tumor response, to evaluate incidence of anti-patritumab antibody, and to explore patritumab-related biomarkers. Patritumab Process 2 formulation was administered as a 60-min intravenous (i.v.) infusion at 18 mg/kg for the initial dose and at 9 mg/kg for the second and subsequent doses every 3 weeks in combination with an oral daily dose of erlotinib 150 mg (150 mg QD). Patients received fixed dose of patritumab Process 2 formulation at 9 mg/kg every 3 weeks after initiation of 18 mg/kg loading dose combined with erlotinib at 150 mg QD in single dose cohort until any of the withdrawal criteria are met. Withdrawal criteria are disease progression, unacceptable toxicity, ILD, dosing postponed/discontinued for more than 3 weeks, subject’s request to withdraw from study treatment, and other instances in which the study cannot be continued in the judgment of the investigator. The initial 21 days after the first administration (cycle 1) were regarded as the DLT evaluation period, and six patients were enrolled at single dose cohort. In the DLT assessments, if none/one of the six patients had a DLT that dose was considered to be tolerable. Adverse events (AEs) were graded using the National Cancer Institute Common Terminology Criteria for AEs, version 4.0. A DLT was defined as any of the following events occurring during cycle 1 (the initial 21 days) as related to either patritumab or erlotinib: (1) grade 3 or higher febrile neutropenia, or persistent (more than 7 days) grade 4 neutropenia; (2) grade 4 thrombocytopenia, or grade 3 thrombocytopenia requiring blood transfusion; (3) uncontrollable grade 3 or higher fatigue, anorexia, nausea, vomiting, skin disorder (e.g., skin eruption, urticaria), and diarrhea despite maximal supportive therapy; (4) grade 3 or higher toxicity, with the exception of (1)–(3) as well as pyrexia without neutropenia, transient electrolyte abnormality, and transient lab-oratory abnormality not requiring treatment and without clinical symptoms; and (5) toxicity requiring suspension of erlotinib therapy for more than 7 days during the DLT evaluation period. Tumor response was determined for all patients with measurable and/or non-measurable lesions according to Response Evaluation Criteria in Solid Tumors (RECIST version 1.1). Tumor measurements by CT or MRI were obtained at baseline, every 6 weeks thereafter and Cycle 1 Day 21.
Pharmacokinetics
Pharmacokinetics were evaluated in all patients received patritumab Process 2 formulation in combination with erlotinib. Blood samples were collected at pre-dose and 1 (end of infusion), 4, 7, 24, and 72 h after the start of first dose infusion, on Days 8 and 15 of Cycle 1, and on Day 1 of Cycles 2, 3, and 4. Serum concentrations of patritumab Process 2 formulation were determined by enzyme-linked immunosorbent assay (ELISA). Pharmacokinetic parameters after the first dose were calculated by non-compartmental analysis using WinNonlin (Ver.6.2 CERTARA G.K., Japan).
Pharmacokinetic statistical analyses were performed using SAS System Release 9.2 (SAS Institute Japan Ltd., Tokyo, Japan).
Biomarkers
Serum soluble HER3 and the HER3 ligand, heregulin (HRG) levels were also evaluated in all patients. Blood for serum biomarkers was collected on Day 1 (before administration), 8, 15, and 21 of Cycle 1, and Day 21 of Cycle 2 and changes in both soluble HER3 and HRG serum levels were evaluated. Soluble HER3 levels were measured by enzyme-linked immunosorbent assay (ELISA). In addition, soluble HRG was also evaluated immunologically according to the previous report [24, 25].
Statistical method
All patients who received study medication were included in the analysis of safety and efficacy. Safety and efficacy statistical analyses were performed by SAS System Release 9.2 (SAS Institute Inc., Cary, NC, USA).
Results
Patient characteristics
Six Japanese patients with advanced NSCLC were enrolled and were evaluated in this study. The baseline characteristics of the patients are summarized in Table 1. The age range was 54.0–78.0 years (median: 72.5 years), and all six patients harbored EGFR mutation (exon 19 deletion, n = 2; L858R, n = 4; T790M, n = 1). Five patients received prior EGFR-TKI therapies treated by at least any one of gefitinib, erlotinib, and afatinib, and one patient was an EGFR-TKI therapy naïve patient. The median number of prior chemotherapy regimens was 2 (range, 1 − 5). At the time of data cutoff, all patients had discontinued treatment: one because of an adverse event (grade 3 ILD), one because of patient withdrawal and four because of disease progression.
Safety and tolerability
The DLTs occurring during cycle 1 were evaluated. No DLTs were observed in all patients. The AEs reported for all treatment cycles are summarized in Table 2.
The most common overall AEs (≥ 50 %) were diarrhea, stomatitis, dermatitis acneiform, dry skin, and paronychia which were generally mild and manageable. Most of the AEs were related to both patritumab and erlotinib and were generally mild and manageable. No grade 4 or grade 5 AEs occurred in this study. One patient discontinued both patritumab and erlotinib treatment because of a drug-related AE. An elderly female patient with gefitinib-pretreated advanced NSCLC developed grade 3 ILD (onset, day 86); after the discontinuation of both patritumab and erlotinib treatment, steroid administration was conducted and ILD was resolving. No patients developed anti-patritumab antibodies after the administration of patritumab Process 2 formulation in this study.
Pharmacokinetics
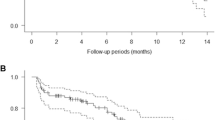
Mean +/− sd serum concentration for patritumab Process 2 formulation versus time is shown in Fig. 1, and descriptive statistics for the PK parameters are summarized in Table 3. The mean area under the curve (AUC) value was 2640 µg day/mL; the Cmax value was 434 µg/mL; and the terminal half-live was 9.18 days, respectively.

The mean ± sd serum concentration for patritumab Process 2 formulation versus time
Efficacy
One partial response (PR) and five cases with stable disease (SD) were observed. The PR was observed in an EGFR-TKI naïve patient who had a tumor with an EGFR-activating mutation (L858R). Among the five SD patients, all patients received prior EGFR-TKIs (treated by at least any one of gefitinib, afatinib, and erlotinib) treatment (exon 19 deletion, n = 2; L858R, n = 3, T790M, n = 1). The median progression-free survival (PFS) (95 % confidence interval) was 222.0 (100.0–363.0) days.
Biomarker analysis (serum HER3 and heregulin)
Soluble serum HRG level was detectable in only a patient, subject No. 2, prior the administration (5,190.1 pg/ml), although was not detectable in other samples from five patients (Table 4). In addition, this patient maintained a most durable stable disease for 363 days among six patients, although tumor already acquired a resistance to gefitinib. The soluble HRG level was maintained on during Cycles 1 and 2 in a patient subject No. 2 (7411.2 pg/ml at Day 8 of Cycle 1; 6870.5 pg/ml at Day 15 of Cycle 1; 5399.1 pg/ml at Day 21 of Cycle 1; and 5329.5 pg/ml at Day 21 of Cycle 2). Soluble HER3 level was various in serum obtained from six patients prior the administration (mean 7,475.8 pg/ml; 3,164.0–25,005.5 pg/ml, Table 4). Soluble HER3 level increased post the administration (mean 15,585.8 pg/ml at Day 8 of Cycle 1; 18,016.7 pg/ml at Day 15 of Cycle 1; 18,100.1 pg/ml at Day 21 of Cycle 1, Supple. Table 2).
Discussion
This phase 1 study was conducted primarily to evaluate the safety and tolerability of patritumab Process 2 formulation combined with EGFR-TKI, erlotinib in Japanese patients with advanced NSCLC. The safety and tolerability, PK, anti-patritumab antibody, tumor response, and biomarkers, including sHER3 and sHRG, were explored in this study. Patritumab Process 2 formulation was developed to manufacture the drug substance with the aim of increasing the yield of the target protein and improving properties of the drug substance and/or product.
Regarding safety and tolerability, no DLTs were reported at all patients (patritumab Process 2 formulation at 9 mg/kg every 3 weeks after initiation of 18 mg/kg loading dose with oral daily dose of erlotinib 150 mg). The most common AEs in this study were gastrointestinal and skin toxicities, which were generally mild and manageable and most AEs in this study were similar to the well-known side effects of EGFR-TKIs. No treatment-related deaths due to AEs were reported. Some SAEs were reported, including grade 3 ILD, which was related to either patritumab or erlotinib treatment. An elder female patient with gefitinib-pretreated advanced NSCLC developed grade 3 ILD (onset, day 86); after the discontinuation of both patritumab and erlotinib treatment, steroid administration was conducted and ILD was resolving.
PK parameters were calculated by non-compartmental model-based analysis, and AUC0−21day and Cmax were compared with existing data of former Process 1 formulation combined with erlotinib in Japanese patients with non-small cell lung cancer. The AUC0−21day and Cmax (Mean ± SD) of the Process 2 formulation were 2640 ± 544 µg day/mL and 434 ± 121 µg/mL, respectively, suggesting almost comparable exposure obtained with Process1 formulation (AUC0−21day and Cmax: 2480 ± 420 µg day/mL and 400 ± 46.7 µg/mL, respectively) [22]. Furthermore, no neutralizing antibodies were detected in patients in this study after patritumab Process 2 formulation administration, as assessed by an anti-patritumab antibody and cell-based bioassay, similar to findings in the previous studies.
In regard to the clinical efficacy of the combined treatment, 1 PR and 5 cases with SD were observed among six patients. The PR patient had a tumor with an EGFR-activating mutation (L858R) and EGFR-TKI naïve setting. Among six patients, five patients had available information about T790M status of their tumor by conducting pre- or post-biopsy of tumor tissue when patients developed disease progression against preceding EGFR-TKIs therapy. Although this was limited patients’ number, one key remarkable point about potential clinical efficacy in this study is that durable progression-free survival was observed (363, 100, and 297 days) in three T790M wild-type EGFR-TKI refractory patients. These results were encouraging, because they were similar or superior to those obtained with use of the third generation EGFR-TKI, osimertinib (AZD9291) in recent clinical study in patients with previously treated NSCLC and sub-population analysis of patients with no detectable EGFR T790M (69 % of the patients had an estimated response duration of 6 months or longer, with a median progression-free survival of 2.8 months (95 % CI, 2.1 to 4.3; 71 % maturity) in 62 patients with no detectable EGFR T790M [26].
The current study observed that a patient had a high level of soluble HRG in serum and maintained most durable stable disease, although tumor already acquired a resistance to gefitinib. Previous studies proved that HRG expression level correlated with the efficacy of several kinds of anti-HER3 antibody preclinically as well as clinically [25, 27–30]. U3-1287 also inhibited cell-proliferation in aberrantly heregulin-expressing NSCLC cells [28]. Furthermore, Patritumab combination with erlotinib demonstrated a significantly improved progression-free survival in patients with advanced NSCLC and high level of soluble HRG in serum compared to placebo combination [25]. Although the current study analyzed limited samples, patritumab plus erlotinib might be optimal in patients with EGFR-mutant NSCLC and high level of HRG expression.
In conclusion, patritumab Process 2 formulation at a dose of 18 mg/kg for the initial dose and at 9 mg/kg for the second and subsequent doses every 3 weeks in combination with an oral daily dose of erlotinib 150 mg was determined to be feasible in regard to the tolerability in Japanese patients with advanced NSCLC. Although some limitations including small patients number exist in this study, preliminary demonstration of both favorable PK profiles and the efficacy of the combined treatment was encouraging, potentially in NSCLC patients with EGFR-activating mutations, where prior EGFR-TKI treatment failed regardless of EGFR T790M status.
References
Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, BenaventeS et al (2008) Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 27:3944–3956
Naidu R, Yadav M, Nair S, Kutty MK (1998) Expression of c-erbB3 protein in primarybreast carcinomas. Br J Cancer 78:1385–1390
Narayan M, Wilken JA, Harris LN, Baron AT, Kimbler KD, Maihle NJ (2009) Trastuzumab-induced her reprogramming in “resistant” breast carcinoma cells. Cancer Res 69:2191–2194
Friess H, Yamanaka Y, Kobrin MS, Do DA, Buchler MW, Korc M (1995) Enhanced erbB-3 expression in human pancreatic cancer correlates with tumor progression. Clin Cancer Res 1:1413–1420
Müller-Tidow C, Diederichs S, Bulk E, Pohle T, Steffen B, Schwäble J et al (2005) Iden-tification of metastasis-associated receptor tyrosine kinases in non-small cell lung cancer. Cancer Res 65:1778–1782
Bieche I, Onody P, Tozlu S, Driouch K, Vidaud M, Lidereau R (2003) Prognosticvalue of ERBB family mRNA expression in breast carcinomas. Int J Cancer 106:758–765
Yi ES, Harclerode D, Gondo M, Stephenson M, Brown RW, Younes M et al (1997) Highc-erbB-3 protein expression is associated with shorter survival in advancednon-small cell lung carcinomas. Mod Pathol 10:142–148
Tanner B, Hasenclever D, Stern K, Schormann W, Bezler M, Hermes M et al (2006) ErbB-3 predicts survival in ovarian cancer. J Clin Oncol 24:4317–4323
Pinkas-Kramarski R, Soussan L, Waterman H, Levkowitz G, Alroy I, Klapper L et al (1996) Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J 15:2452–2467
Campbell MR, Amin D, Moasser MM (2010) HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin Cancer Res 16:1373–1383
Xia W, Gerard CM, Liu L et al (2005) Combining lapatinib (GW572016), a small molecule inhibitor of ErbB1 and ErbB2 tyrosine kinases, with therapeutic anti-ErbB2 antibodies enhances apoptosis of ErbB2-overexpressing breast cancer cells. Oncogene 24:6213–6221
Sergina NV, Rausch M, Wang D et al (2007) Escape from HERfamily tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 445:437–441
Jain A, Penuel E, Mink S, Schmidt J et al (2010) HER kinase axis receptor dimer partner switching occurs in response to EGFR tyrosine kinase inhibition despite failure to block cellular proliferation. Cancer Res 70:1989–1999
Wang S, Huang X, Lee C-K, Liu B (2010) Elevated expression of erbB3 confers paclitaxel resistance in erbB2-overexpressing breast cancer cells via upregulation of Survivin. Oncogene 29:4225–4236
E ngelman JA, Zejnullahu K, Mitsudomi T, Song Y et al (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316:1039–1043
T reder M, Hartmann S, Ogbagabriel S, Borges E et al (2008) Fully human anti-HER3 monoclonal antibodies (mAbs) inhibit oncogenic signaling and tumor cell growth in vitro and in vivo. In: 99th AACR annual meeting (meeting abstract): LB-20, San Diego, CA
Freeman D, Ogbagabriel S, Rothe M, Radinsky R, Treder M (2008) Fully human anti-HER3 monoclonal antibodies (mAbs) have unique in vitro and in vivo functional and antitumor activities versus other HER family inhibitors. In: 99th AACR annual meeting (meeting abstract): LB-21, San Diego, CA
Treder M, Ogbagabriel S, Moor R, Schulze-Horsel U, Hettmann T, Rothe M et al (2008) Fully human anti-HER3 mAb U3-1287(AMG 888) demonstrates unique in vitro and in vivo activities versus other HER family inhibitors in NSCLC models. Eur J Cancer 6(suppl):99(abstract 309)
Freeman DJ, Ogbagabriel S, Bready J, Sun J-R, Radinsky R, Hettmann T (2011) U3–1287 (AMG 888), a fully human anti-HER3 mAb, demonstrates in vitro and in vivo efficacy in the FaDu model of human squamous cell carcinoma of the head and neck (SCCHN). Mol Cancer Ther 10(suppl 1):(abstract A182)
LoRusso P, Jänne PA, Oliveira M, Rizvi N, Malburg L, Keedy V et al (2013) Phase I study of U3-1287, a fully human anti-HER3 monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res 19:3078–3087
Wakui H, Yamamoto N, Nakamichi S, Tamura Y, Nokihara H, Yamada Y et al (2014) Phase 1 and dose-finding study of patritumab (U3-1287), a human monoclonal antibody targeting HER3, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 73:511–516
Nishio M, Horiike A, Murakami H, Yamamoto N, Kaneda H, Nakagawa K, Horinouchi H, Nagashima M, Sekiguchi M, Tamura T (2015) Phase I study of the HER3-targeted antibody patritumab (U3-1287) combined with erlotinib in Japanese patients with non-small cell lung cancer. Lung Cancer 88(3):275–281
Mukai H, Saeki T, Aogi K, Naito Y, Matsubara N, Shigekawa T, Ueda S, Takashima S, Hara F, Yamashita T, Ohwada S, Sasaki Y (2016). Patritumab plus trastuzumab and paclitaxel in human epidermal growth factor receptor 2-overexpressing metastatic breast cancer. Cancer Sci. Jul 25. [Epub ahead of print]
Kawakami H, Yonesaka K (2016). HER3 and its ligand, heregulin, as targets for cancer therapy. Recent Pat Anticancer Drug Discov. Apr 18. [Epub ahead of print]
Yonesaka K, Hirotani K, von Pawel J, Dediu M, Chen S, Copigneaux C, et al (2016). Soluble heregulin, HER3 ligand, to predict the efficacy of anti-HER3 antibody patritumab combination with erlotinib in randomized phase II study, HERALD, for non-small cell lung cancer. J Clin Oncol 34 (suppl; abstr 9071)
Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, Frewer P, Cantarini M, Brown KH, Dickinson PA, Ghiorghiu S, Ranson M (2015). AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer Engl N J Med. 372(18):1689–99
Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M et al (2010) An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res 70:2485–2494
Yonesaka K, Hirotani K, Kawakami H, Takeda M, Kaneda H, Sakai K et al (2016) Anti-HER3 monoclonal antibody patritumab sensitizes refractory non-small cell lung cancer to the epidermal growth factor receptor inhibitor erlotinib. Oncogene 35(7):878–886
Garner AP, Bialucha CU, Sprague ER, Garrett JT, Sheng Q, Li S et al (2013) An antibody that locks HER3 in the inactive conformation inhibits tumor growth driven by HER2 or Neuregulin. Cancer Res 73(19):6024–6035
Sequist LV, Lopez-Chavez A, Doebele RC, Gray JE, Harb WA, Modiano MR, et al (2014). A randomized phase 2 trial of MM-121, a fully human monoclonal antibody targeting ErbB3, in combination with erlotinib in EGFR wild-type NSCLC patients. J Clin Oncol 32:5 s(suppl; abstr 8051)
Acknowledgements
Research funding was provided by Daiichi Sankyo Co., Ltd. (Tokyo, Japan).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors have no potential conflicts of interest to report.
Ethical standards
The study was performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. The study was approved by the institutional review board of each study site, and all the patients provided informed consent prior to their inclusion in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shimizu, T., Yonesaka, K., Hayashi, H. et al. Phase 1 study of new formulation of patritumab (U3-1287) Process 2, a fully human anti-HER3 monoclonal antibody in combination with erlotinib in Japanese patients with advanced non-small cell lung cancer. Cancer Chemother Pharmacol 79, 489–495 (2017). https://doi.org/10.1007/s00280-016-3231-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-016-3231-3