
Abstract
In October 2019, a viral infectious disease appeared in the city of Wuhan in China. A new betacoronavirus, SARS-CoV-2, has been recognized as the responsible pathogen in this infection. Although coronavirus disease is principally expressed as a pulmonary infection, critical SARS-CoV-2 infection is frequently complicated with coagulopathy, and thromboembolic events are recognizable in several patients. Dehydration, acute inflammatory condition, protracted immobilization during disease, existence of multiple cardiovascular risk factors such as diabetes, obesity or hypertension, previous coronary artery disease, ischemic stroke, peripheral artery disease are frequent comorbidities in SARS-CoV-2 hospitalized subjects, which possibly augment thrombo-embolic risk. However, other causal factors can still be identified such as unrestricted angiotensin II action, the use of immunoglobulins, an increased production of adhesion molecules able to induce vascular inflammation and endothelial activation, complement stimulation, excessive production of neutrophil extracellular traps (NETs), and increased platelet count. Low-molecular-weight heparin should be chosen as early treatment because of its anti-inflammatory action and its ability to antagonize histones and so defend the endothelium. However, several therapeutic possibilities have also been proposed such as fibrinolytic treatment, drugs that target NETs, and complement inhibition. Nevertheless, although the violence of the pandemic may suggest the use of heroic treatments to reduce the frightening mortality that accompanies SARS-CoV-2 infection, we believe that experimental treatments should only be used within approved and controlled protocols, the only ones that can provide useful and specify information on the validity of the treatments.
Similar content being viewed by others
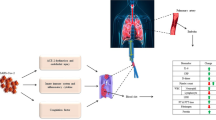


Introduction
Clinical pattern and laboratory findings of thromboembolic events in SARS-CoV-2 patients
In October 2019, a viral infectious disease appeared in the city of Wuhan in Hubei Province, China. A new betacoronavirus, SARS-CoV-2, able of human-to-human diffusion, has been recognized as the responsible pathogen in this infection [1, 2]. At the time of writing, the global pandemic is still present. Latest balance calculates > 8,000,000 people affected worldwide, with > 450,000 deaths.
Although it is well known that coronavirus disease 2019 (COVID-19) is principally expressed as a pulmonary infection, several findings suggest that it should be considered as a systemic pathology implicating several organs and systems comprising neurological, cardiovascular, gastrointestinal, hematopoietic, and immune system [3,4,5]. Furthermore, as reported by numerous research, grave SARS-CoV-2 infection is frequently complicated with coagulopathy, and thromboembolic events are recognizable in several patients [6, 7]. In a retrospective study, 260 out of 560 subjects (46.4%) with laboratory proved SARS-CoV-2 disease had an increase of D-dimer, and the augment was more prominent among grave patients (59.6% vs 43.2%). Authors argue that D-dimer alteration can indicate the gravity of the infection and an augmented concentration is correlated with a poorer prognosis [8].
These results were confirmed by other studies. A different retrospective analysis performed in China comprising 41 subjects demonstrated that prothrombin time (PT) and D-dimer concentrations were greater on admittance in infected subjects necessitating Intensive Care Unit (ICU) assistance (median PT 12.2 s for intensive care vs 10.7 s; median D-dimer 2.4 mg/L for intensive care assistance vs 0.5 mg/L for non-intensive care assistance), whereas augmented D-dimer concentrations were also connected with death in the multivariable analysis [9, 10].
In the analysis performed by Tang et al., including information from 183 subjects with SARS-CoV-2 disease, on admittance, patients who died had substantively greater fibrin degradation products (FDP) concentrations and augmented PT and activated partial thromboplastin time (aPTT) with respect to survivors, with a reduction of fibrinogen and antithrombin (AT III) levels [11]. Remarkably, 71.4% of patients who died vs 0.6% of patients who survived satisfied the criteria for disseminated intravascular coagulation (DIC). In a prospective research valuing D-dimer and FDP concentrations in SARS-CoV-2 patients and normal subjects, infected patients presented greater concentrations of these parameters, and patients with more serious disease presented greater values of FDP and D-dimer with respect to patients with minor symptoms [12]. The average time interval from admittance to DIC onset was 4 days.
Thus, DIC emerged in most of the deaths, and this is not unexpected as infection is one of the most frequent causes of DIC. It is well-known that DIC starts when endothelial cells and monocytes are stimulated to produce cytokines after a serious damage, with abnormal production of von Willebrand factor and Tissue Factor (TF). The subsequent circulation of uninhibited thrombin can stimulate platelets and activate fibrinolysis [13].
In a previous study, Gralinski et al. explored effects of SARS-coronavirus disease on coagulation.
Their results propose that modification of the urokinase pathway during infection causes a graver lung alteration and that plasminogen activator inhibitor-1 (PAI-1) has a defending action after the infection [14]. Moreover, Berri et al. confirmed that plasminogen aggravates the inflammation due to infection, while fibrinolysis may also be provoked by severe infection [15].
Nevertheless, although Tang et al. described findings congruent with a condition of DIC (high FDP and D-dimer, elongated PT and APTT, and reduced platelet counts) [11], only a part of these data (increased D-dimer) have been confirmed by other studies, while other alterations such as increased PT/APTT are not confirmed. Moreover, decreased fibrinogen clotting activity and low platelet counts, which are characteristic elements of DIC, are reported normal or even increased in other studies [16].
The causes for this incongruity are uncertain. It cannot be excluded that the two groups of patients, although both hospitalized to the ICU, were at diverse stages in the course of the infection.
An additional factor of doubt is the fact that it is difficult to differentiate between the laboratory alterations seen in subjects with coagulopathy due to sepsis-caused hepatic dysfunction rather than DIC [17]. This difference is especially important given the findings which propose that hepatitis is present in several subjects with SARS-CoV-2 infection, thus imitating the coagulopathy.
In any case, the clinical expressions of coagulopathy in these subjects can involve the most different organs and systems, from the heart, to the kidney, to the lungs, to the peripheral vessels. Older subjects and those with other diseases are at augmented danger of death from SARS-CoV-2, but younger subjects without other comorbidities may also have fatal problems such as thromboembolic complications [18]. Among other things, with regard to mortality, it should be borne in mind that subjects displaying with cardiac damage in the context of SARS-CoV-2 infection are more susceptible to coagulation alterations with respect to those without cardiac injury [19].
The limited data presented on thrombotic complications in subjects with COVID-19 propose that rates of venous thromboembolic events may be as high as 25 to 30%, especially in critically ill, mechanically ventilated subjects. Thrombotic complications also comprise stroke, acute limb ischemia, and acute coronary syndromes [20,21,22,23].
Several studies stated an augmented occurrence of VTE in subjects admitted to the ICU with SARS-CoV-2 infection. Klok et al. registered a 27% VTE occurrence and 3.7% arterial vascular thrombosis in spite of the employ of standard VTE prophylaxis with low-molecular-weight heparin (LMWH) [24], certainly a higher percentage with respect to the failure rate of 7.7% generally registered for VTE prophylaxis in ICU setting [25]. Cui et al. reported an analogous 25% frequency of VTE, but VTE prophylaxis was not provided in this population as VTE prophylaxis is not habitually used in Asia [26, 27].
SARS-CoV-2: pathogenetic mechanisms of coagulation alterations
Dehydration, an acute inflammatory condition, protracted immobilization during disease, existence of multiple cardiovascular risk factors such as diabetes, obesity or hypertension, previous coronary artery disease, ischemic stroke, peripheral artery disease, or VTE and classical genetic thrombophilia, such as heterozygous factor II and factor V Leiden mutation are frequent comorbidities in SARS-CoV-2 hospitalized subjects, which possibly augment VTE risk.
However, other causal factors can still be identified (Table 1).
Inflammatory mediators
SARS-CoV-2 presents several correspondences with the SARS virus; nevertheless, SARS-CoV-2 has developed numerous characteristics that make it a more effectual agent for contagion than SARS-CoV. The most important receptor-binding domain of SARS-CoV-2 preserved the overall structure of the SARS-CoV binding domain, comprising 8 of the 14 residues being totally the same. However, the 3D conformation of the SARS-CoV-2 binding site displays that it is more compacted, has ameliorated binding ability, and possibly increased ACE-2 receptor binding affinity [66,67,68,69].
Additional distinction is that SARS-Cov-2 includes a polybasic (furin) cleavage site introduced at the boundary of the S1/S2 subunits of the spike S-protein12 able to increase the virus’ capacity to internalize into the human cells.
After virus contact with the ACE-2 receptor in the presence of TMPRSS2, SARS-CoV-2 can go into the cells via endocytosis or membrane fusion [70,71,72]. Experimental model demonstrated that after engagement of SARS-CoV virus with ACE-2 receptor, there is an important decrease of ACE-2 in the endothelial cells [73]. This is probably part of the protection systems in response to the infection, to reduce viral growth and diffusion. Nevertheless, the possible effect of this engagement is that the physiological action normally performed by the receptor is also considerably reduced. This can cause unrestricted angiotensin II actions, comprising prothrombotic, proinflammatory, and prooxidant effects. Moreover, the onset of an endothelial damage due to the virus attachment to ACE-2 receptor may further augment VTE risk, while the delivery of inflammatory mediators and the use of drugs such as steroids and immunoglobulins in critical subjects may provoke an augmented blood viscosity [59, 60].
Numerous inflammatory mediators are involved in coagulative dysregulation during acute infection. Firstly, tissue factor (TF), the primary initiator of the blood coagulation cascade, provokes rapid hemostasis in case of organ injury through its receptor activity for factor VII. Inflammatory cytokines, such as tumor necrosis factor (TNF)-α, and endotoxin potently stimulate expression of TF on the cell surface of endothelium and leukocytes, specifically monocytes. TF stimulates pleiotropic inflammatory responses and gives a powerful contribution to hypercoagulable state [55].
Stimulation of cells like macrophages can also cause the delivery of cytokines, comprising IL-1β and IL-6, which will increase production of adhesion molecules able to induce vascular inflammation and endothelial activation [28]. With the decrease of ACE-2 and increase of angiotensin II, the generation of PAI-1 is also increased. This event causes an increased vascular inflammation and aggravates the prothrombotic condition. All these findings have been found in subjects with serious infection and laboratory evidence of augmented D-dimer and IL-6 concentrations.
Deficient coagulation inhibitors
Some findings propose that subjects with SARS-CoV-2 infection may acquire a condition of hypercoagulability as demonstrated by the thromboelastographic parameters, augmented factor VIII, fibrinogen, and von Willebrand factor. This condition could promote the onset of pulmonary embolism or deep vein thrombosis of the lower limbs in infected patients.
The motives for this hypercoagulability are unclear. In this type of patients, factor VIII, which is one of the most powerful stimulants of hypercoagulability, is intensely augmented, and the principal naturally occurring anticoagulants are normal (AT III) or even augmented (protein C) [16].
Moreover, damage of endothelial cells caused by SARS-CoV-2 provokes an augmented thrombin production and fibrinolysis block [56, 57].
Acute fibrinolysis shutdown has been reported in early sepsis and found to correlate to augmented morbidity and mortality. Fibrinolysis shutdown and organ failure have historically been associated with DIC [57].
In a recent report, fibrinolysis shutdown, as demonstrated by augmented D-dimer and complete failure of clot lysis at 30 min on thromboelastography, predicts thromboembolic complications and need for hemodialysis in critically ill patients with SARS-CoV-2 [58].
However, there are several limitations to this retrospective descriptive study. First, there was variability in the laboratory testing patterns based on intensivist preference. In addition, coagulation parameters were drawn at variable times of the patient disease processes. Studies done to evaluate for thromboembolic events were performed for clinical suspicion rather than routine screening, and therefore some events were likely not captured or had delays in diagnosis.
Furthermore, hypoxia can also induce thrombosis via not only augmenting blood viscosity but also via a hypoxia-inducible transcription factor-dependent system [65]. Hypoxia could be secondary to pneumonia, ARDS, hyperthermia (which may activate platelets and coagulation), and hypovolemia (due to gastrointestinal fluid loss). Vascular response to hypoxia is regulated essentially by the hypoxia-inducible transcription factors (HIFs); HIFs may stimulate further inflammation, thus augmenting blood viscosity and contributing to worsen hypercoagulability. Moreover, HIFs may directly activate platelets and coagulation factors, augmenting TF expression, augmenting PAI-1, and inhibiting the endogenous anticoagulant proteins [30].
As pointed out above, the action of SARS-CoV-2 infection on coagulation and fibrinolysis is believed to be controlled by several proinflammatory cytokines, similarly to what happens in cases of pneumonia due to other pathogens [74, 75]. The model of thrombosis is the pattern of interaction between coagulation and inflammatory state. All the subjects with coagulation alterations presented increased levels of IL6, and an evident correlation between IL6 and fibrinogen concentrations was reported. In fact, IL6 is a potent proinflammatory element, which stimulates fibrinogen and platelet production and tissue factor (TF) gene expression in different type of cells such as monocytes and endothelial. TF stimulates thrombin production, and all these elements cause the generation of a procoagulant profile that was evident in SARS-CoV-2 patients [76, 77].
Complement activation
However, numerous clinical and experimental data allow to hypothesize other mechanisms capable of conditioning the onset of thromboembolic events. In fact, more precise indications on the pathogenetic moments of thrombotic events of SARS-CoV-2 subjects arose from the autopsy findings of these subjects.
Dolhnikoff et al. have settled a system for executing ultrasound-based minimally invasive autopsies, and they used it in mortal cases of SARS-CoV-2 disease to define the pathogenesis of this infection. The pulmonary histological feature is an exudative and proliferative diffuse alveolar injury, with severe epithelial viral cytotoxic signs implicating alveolar and small airway epithelium, and the presence of a small lymphocytic penetration. Moreover, they detected a changing amount of small fibrinous thrombi in pulmonary arterioles. The presence of a great quantity of pulmonary megakaryocytes in the pulmonary capillaries are other signs of stimulation of the coagulation system. Their anatomo-pathological findings corroborate the present perception of hypercoagulative condition in these critically ill subjects [78].
However, surprising data come from analysis conducted by other authors, albeit on a small number of cases. Magro et al. studied pulmonary tissues from subjects with grave SARS-CoV-2 infection and respiratory failure. The feature of SARS-CoV-2 pneumonitis was essentially a pauci-inflammatory septal capillary damage with relevant septal capillary luminal and mural fibrin accumulation. Yet, the most interesting aspect of their analysis was the discovery of relevant accumulations of terminal complement components C5b-9, C4d, and mannose-binding lectin-associated serine protease 2, in the micro-vessels, coherent with continuous, generalized stimulation of the alternative and lectin-based complement pathways. Furthermore, they found a co-localization of SARS-CoV-2 spike glycoproteins with C4d and C5b-9 in the interalveolar septa [79].
The complement system is a component of the innate immune response that protects against viral infections. Two experimental animal studies evaluated complement stimulation in coronavirus infections. In a murine model, animals were missing C3 and thus incapable to stimulate the common complement pathway. In this group, SARS-CoV infection gravity was reduced with less pulmonary alteration and inferior cytokine concentrations in spite of identical viral loads. In a different murine model of MERS-CoV infection, augmented levels of C5a and C5b-9 were reported in pulmonary tissues. Stopping C5a with a murine antibody reduced pulmonary injury with diminished cytokine response and viral proliferation [80, 81].
Confirmation for the participation of the lectin pathway (LP) in SARS-CoV-induced alterations derives from the finding that mannose-binding lectin connects with the SARS-CoV spike glycoprotein [82,83,84]. Even though reported experimentally only for SARS-CoV, with possible limitations regarding the possibility of applying this concept in the current pandemic [85], glycosylation sites for high-mannose elements with the ability to similarly engage mannose-binding lectin have been also recognized for SARS-CoV-2 [70].
Disproportionate complement activation happens in several pathologic conditions, provoking a systemic thrombotic microangiopathy (TMA). Atypical hemolytic uremic syndrome (aHUS) is an infrequent disease of uninhibited complement stimulation followed by microangiopathic hemolytic anemia, thrombocytopenia, and acute renal failure [85]. This condition shows numerous similarities with the microangiopathic alterations found in some patients with SARS-CoV-2 infection. Based on these data, albeit limited to a small number of subjects, grave SARS-CoV-2 infection may provoke a catastrophic, complement-mediated thrombotic microvascular damage condition due to the activation of complement pathways and to a concomitant procoagulant condition [86, 87].
Neutrophil extracellular traps
Another interesting aspect detectable by the autopsy data obtained from patients suffering from SARS-CoV-2 infection was the high amount of intra-alveolar and septal neutrophils. SARS-CoV-infected animals also had higher degree of neutrophils in the lung. In humans, tissue neutrophilia may be due to the neutrophil chemoattractant ability of complement [88] and is correlated with a poorer outcome and is associated with the severity of disease [89,90,91]. The neutrophil-to-lymphocyte ratio is also an independent risk factor for more serious disease [9]. Although leukocytosis and neutrophilia are well-known characteristics of acute infection, in SARS-CoV-2 infection, neutrophilia could contribute to the genesis of a prothrombotic condition through the excessive production of neutrophil extracellular traps (NETs) [35].
In fact, neutrophils have a different, less known method of destroying pathogens: the generation of NETs [36]. NETs are complexes of nucleic DNA, histones, and nucleosomes ejected from the neutrophils that entrap pathogens. Crucial elements in the generation of NETs are enzymes such as neutrophil elastase (NE), which damages intracellular proteins and provokes nuclear fragmentation; gasdermin D which provokes pores in the membrane of the neutrophil, thus accelerating cell membrane rupture and the emission of DNA; and peptidyl arginine deiminase type 4 (PAD4), which citrullinates histones to ease the decondensation of the chromosomal DNA [37].
Even though NETs are useful in the protection against pathogens, collateral injury from continuous NET generation also provokes several negative situations, comprising those that happen during viral diseases [38]. Actually, disproportionate NET generation can promote micro-thrombosis, stimulate several inflammatory responses that damage contiguous tissues, and provoke a serious injury to lung, heart, and kidney [39], three organs frequently altered in SARS-CoV-2 infection.
NET may produce procoagulant elements correlated with plasma hypercoagulability and augmented risk of thrombosis in animal and human experimental models [40,41,42].
In fact, increased blood concentrations of NETs may justify hyperactive coagulation retrieved in SARS-CoV-2 subjects, and intravascular NETs have been demonstrated to have a crucial action in onset and progression of thrombotic events in arteries and veins. For example, NET concentrations correlate with thrombin levels, and when NETs are present in great quantities in blood, they can cause the closure of small vessels [43, 44]. In experimental models of septicaemia, intravascular NETs can cause the formation of microthrombi and provoke damage to different organs [45]. Moreover, a correlation between NETs and IL1β exists. If a NET–IL1β loop is activated in severe SARS-CoV-2, the accelerated generation of NETs and IL1β could accelerate respiratory alteration, the formation of microthrombi, and aberrant immune responses.
As far as the intimate mechanisms of action of NETs are concerned, NETs stimulate the plasma kallikrein–kinin system (the contact pathway of coagulation) through electrostatic relationships between the NET histones and platelet phospholipids [46]. Histones can also stimulate platelet activation by operating as ligands for the Toll-like receptors on platelets [47]. Moreover, NE probably also operates by destroying the principal coagulation inhibitors such as tissue factor pathway inhibitor and AT III [48]. Moreover, there is probably a response loop by which thrombin causes platelet stimulation, and stimulated platelets then increase NET generation [33].
In animal models, destroying NETs with DNase I reestablishes normal perfusion of the heart and kidney [49].
A different reason for the hypercoagulability of these patients may be the occurrence of a great amount of circulating microvesicles. These elements are cytoplasmic microparticles derived from platelets or other cells, which contain procoagulant factors such as the TF or phosphatidylserine. Microvesicles are recognized factors of venous thromboembolism [31], while augmented amount of circulating microvesicles have been described in septic subjects [32], and it is consequently probable that they are also augmented in subjects with SARS-CoV-2 infection.
Platelet alteration
Finally, a further reason for hypercoagulability could lie in an alteration in the number of platelets. In SARS-CoV-2-infected patients, it is possible to detect both a reduction and an increase in platelets. A meta-analysis of nine reports has proposed that thrombocytopenia is notably correlated with the gravity of the SARS-CoV-2 infection [51]. Nevertheless, Qu et al. demonstrated that among hospitalized subjects with SARS-CoV-2 infection, those appearing with thrombocytosis had worse prognosis. Furthermore, the platelet-to-lymphocyte ratio at the time of platelet increase appeared as an independent prognostic factor for protracted hospitalization and may suggest a more relevant cytokine storm due to augmented platelet activation [52]. Yin et al. compared the coagulation parameters between SARS-CoV-2 infection and pneumonia provoked by different agents. Subjects with pneumonia caused by SARS-CoV-2 had greater platelet count than those caused by non-SARS-CoV-2 agents [53]. Nevertheless, as organ alteration is principally restricted in the lung, the coagulation aspect of severe SARS-CoV-2 infection might not be the same of the sepsis in general. Due to the augmented production of thrombopoietin subsequent to pulmonary inflammation, platelet number may not be a reliable marker for coagulopathy of SARS-CoV-2 infection [54].
Coagulation alterations in SARS-CoV-2 infection: management strategies
High blood pressure is a known risk factor for complications in patients with SARS-CoV-2. Since ACE-2 is the most important virus receptor, it is worth asking whether some drug classes (i.e., ACE inhibitors, angiotensin II receptor blockers) should be discontinued.
In a recent study conducted in patients with SARS-CoV-2, the viral load and lung injury were strongly associated with the circulating levels of angiotensin II [61].
ACE-2 down-regulation caused by SARS-CoV-2 might be remarkably disadvantageous in subjects with baseline ACE-2 deficiency due, for example, to older age or diabetes, hypertension, and heart disease. A further ACE-2 down-regulation provoked by the infection might increase the dysregulation between the adverse and the protective receptor axis, with augmented progression of inflammatory and thrombotic processes caused by angiotensin II hyperactivity unopposed by angiotensin [62], and numerous experimental models of lung injury caused by diverse triggers documented the causative relation between down-regulation of ACE-2 receptors and pulmonary inflammation provoked by an imbalance between angiotensin II over-activity and of antiotensin1–7 deficiency [63].
If we assume that viral-provoked ACE-2 deficiency and the resulting angiotensin II over-activity unopposed by angiotensin1–7 are relevant in the progression of the disease, then recombinant ACE-2, angiotensin1–7, and angiotensin II type 1 receptor blockers might be rational approaches in these subjects [92,93,94].
Moreover, based on what has been reported above on the pathophysiological mechanisms that regulate the state of hypercoagulation and thrombotic complications of patients suffering from SARS-CoV-2 infection, various therapeutic possibilities have been proposed (Table 2).
As far DIC, to date, its therapy has been direct on treatments to cure the principal-related disease [113]; this, of course, is inadequate in the case of SARS-CoV-2 infection, until more is discovered about efficient antiviral drugs for this new disease. However, the International Society of Thrombosis and Haemostasis has suggested a novel class of patients recognizing an earlier stage of sepsis-related DIC, named “sepsis-induced coagulopathy,” and these subjects benefit from anticoagulant treatment [114].
Low-molecular-weight heparin (LMWH), or unfractionated heparin (UFH) should be chosen over direct oral anticoagulants for the potential interactions with antiviral and antibacterial (such as azithromycin) therapy [95].
LMWH is the most frequently employed anticoagulant also because of its anti-inflammatory action [96]. In fact, one of the better recognized non-anticoagulant capabilities of heparin, its anti-inflammatory effect, may also be crucial in this group of patients. The mechanisms of the anti-inflammatory action of heparin comprise blocking neutrophil chemotaxis and leukocyte migration, connecting to inflammatory cytokines, counterbalancing the positively charged peptide complement factor C5a, and reducing acute phase proteins [97,98,99]. A recent report stated that heparin can reduce the concentration of inflammatory biomarkers [100].
In addition to the pathogenic invasion, able to cause endothelial alterations, histones discharged from injured cells can also provoke endothelial damage [29]. Heparin can antagonize histones and so defend the endothelium [101, 102]. This protecting action can be expanded to the endothelial tight junctions as established in a sepsis model, where heparin reduced vascular leakage subsequent lipopolysaccharide-induced damage [103]. A different protective mechanism is via its action on histone methylation and the NF-κB and MAPK pathways [104]. Thus, heparin can influence the microcirculatory alteration and probably reduce organ injury.
If the employ of heparin seems to be the most correct therapeutic choice, various considerations must be made on the dosage to be used.
Rannuci et al. described coagulation parameters in SARS-CoV-2 subjects with acute respiratory distress syndrome. Patients received 4000 IU nadroparin twice daily. First findings were analogous to other studies, with an increased fibrinogen. However, in spite of normal clotting times on viscoelastic testing, other parameters revealed augmented clot firmness with above normal values for contributions from fibrinogen and platelets. For this reason, nadroparin dosing was augmented to 6000 IU or 8000 IU BID if BMI > 35 kg/m2 [115]. The employ of an augmented dosage of nadroparin caused a relevant reduction in D-dimer concentrations even if all subjects still had levels above the normal range. Moreover, viscoelastic testing revealed reduced hypercoagulability after augmenting LMWH and diminutions in clot firmness as a result of a reduced contribution from fibrinogen and platelets.
In SARS-CoV-2 subjects, a risk-adapted method to increase the dosage of heparin should be carefully evaluated. Monitoring renal function and coagulation parameters should be performed.
Furthermore, as genetic risk factors of VTE differ considerably among ethnic populations, and the occurrence of VTE in Asian populations is smallest [116], a greater dose of LMWH could be administered in non-Asian subjects with grave SARS-CoV-2 infection.
Moreover, one potential challenge in the use of unfractionated heparin is the utility of the aPTT for monitoring heparin. In subjects with SARS-CoV-2, besides the intensity of heparin-based regimens, substantial heterogeneity in the aPTT response may be driven by high concentrations of factor VIII and fibrinogen or the presence of a lupus anticoagulant [34, 117]. Consequently, anti-factor Xa levels may need to be measured to ensure that a therapeutic heparin level is achieved [118].
In any case, the efficacy of anticoagulant treatment for sepsis-associated DIC is still debateable [119,120,121], although some reports proposed that septic subjects might just profit from early diagnosis and specific therapy [122, 123].
Caution should be also used for the employ of heparin in DIC associated with SARS-CoV-2 in subjects in treatment with veno-venous extracorporeal membrane oxygenation (VV-ECMO). In fact, an increased rate of intracranial hemorrhage in subjects receiving VV-ECMO was described [124, 125].
Finally, the stimulation of coagulation also provides compartmentalization of pathogens and decreases their diffusion [105]. So, in infected subjects without relevant coagulopathy, anticoagulant therapy could lead to an increased risk of complications. This may justify the greater mortality of heparin users with respect to non-users in subjects with D-dimer ≤ 1 ULN, even though the difference was not statistically relevant.
In any case, augmenting heparin dosage alone is likely inadequate to modify prognosis in subjects with critical severe SARS-CoV-2 acute lung damage. Additional treatment with diverse drugs may be essential to reduce the thrombo-inflammatory condition and successive tissue damages that cause the prothrombotic modification leading to microvascular thrombosis or manifest VTE.
However, it should be noted that none of the therapeutic possibilities listed below have been validated by extensive controlled studies, and therefore the possibility of their use in the clinical setting should be considered only as a proposal to be explored.
A therapeutic possibility could be the concomitant administration of heparin and thrombolytic drugs in subjects with pulmonary embolism. Acute respiratory distress syndrome has been correlated with microclots in the lungs, while elimination of the microclots can be attained by stimulation of endogenous plasmin. In an old experimental animal model, Hardaway et al. evaluated fibrinolytic treatment in acute respiratory distress syndrome, achieving good results [106].
An opportunity could be the employ of bolus doses of alteplase (tPA) while subjects continue the therapeutic heparin therapy. Such an attempt employing greater bolus-dose tPA (100 mg) without maintaining anticoagulation is a different possibility, as mortality in SARS-CoV-2 acute respiratory distress syndrome is extremely high, while the risk of catastrophic bleeding from tPA in non-stroke subjects is about 1% [107].
A diverse chance could be the dipyridamole, a phosphodiesterase inhibitor that blocks platelet aggregation by augmenting intracellular levels of cyclic adenosine monophosphate. Dipyridamole has antiviral actions in vitro, specifically confirming the affinity of dipyridamole for a SARS-CoV-2 main protease [126]. In mouse models of viral pneumonia, dipyridamole administration stimulated interferon response and increased survival in infected mice.
To date, one study has examined dipyridamole in the therapy of SARS-CoV-2; 31 patients with viral infection were randomized to dipyridamole (150 mg three times a day for 7 days) versus control. In this small study, those treated with dipyridamole showed trends toward higher cure and hospital discharge rates. Augmented platelet counts and reduced D-dimer concentrations were also noted with dipyridamole treatment, attributed to infection resolution [64, 127].
Moreover, beyond their anticoagulant actions, direct oral anticoagulants (DOACs), especially factor Xa inhibitors, may exert anti-inflammatory actions in SARS-CoV-2. There is currently one registered clinical trial (C-19-ACS) evaluating low-dose rivaroxaban along with dual antiplatelet therapy, statins, and a proton-pump inhibitor in subjects with SARS-CoV-2 and a suspected acute coronary syndrome (NCT04333407) [128].
As reported above, the studies performed on critical SARS-CoV-2 patients revealed disproportionate complement stimulation that was accompanied by increased D-dimer, bilirubin and LDH, reduction of Hb and platelet count, cardiac and renal damage, and diffuse thrombotic microangiopathy. For this reason, complement inhibition may be a novel possibility in curing SARS-CoV-2 systemic thrombosis [50].
Finally, substances that target NETs are present or are in study. These substances comprise inhibitors of the enzymes or molecules essential for NET generation such as NE, PAD4, and gasdermin D. For instance, endogenous inhibitors of NET generation have been separated from umbilical cord plasma [108], and these could be used for the therapy of inflammatory diseases such as SARS-CoV-2 infection. The NET inhibitor sivelestat was accepted to treat ARDS, but it did not increase survival after ARDS in a meta-analysis of clinical trials [35], while a new group of powerful NE inhibitors, comprising elafil, alvelestat, lonodelestat (POL6014), and CHF6333, have started phase I testing.
DNase proteins, such as DNase 1–like 3, which is being developed to dissolve NETs [109], could enter clinical development soon. Dornase alfa is generally administered through nebulizers, but in several medical centers, these are precluded in SARS-CoV-2 due to the risk of aerosolizing virus and endangering healthcare workers. However, approaches exist that deliver aerosols in closed circuits for mechanically ventilated subjects. For non-intubated subjects, treatment can be safely nebulized in negative pressure rooms. In addition to their possible actions on mucous secretions, DNase treatments may also prevent the further progression to ARDS, as DNase I delivered through the airways increases survival in relevant animal models [110, 111].
In addition to directly targeting NETs, the NET–IL1β loop could be antagonized with approved drugs against IL1β, such as anakinra, canakinumab, and rilonacept [112].
Finally, recent reports indicated that the corticosteroid dexamethasone may reduce mortality of severe COVID-19 patients [129]. Dexamethasone would limit the production of and damaging effect of the cytokines but will also inhibit the protective function of T cells and block B cells from making antibodies, potentially leading to increased plasma viral load [130]. Probably, dexamethasone may be useful for the short term in severe, intubated, COVID-19 patients.
In conclusion, in spite of the evidence of a correlation between inflammation and coagulative alterations, no certain evidence is available of the effectiveness and safety of heparin or antiplatelet substances or other drugs on SARS-CoV-2 subjects, and several problems have to be resolved, such as the correct timing, doses, and associations.
Despite the uncertainties mentioned above, it is certainly possible to try to provide some indications for clinical practice. The employ of LMWH or fondaparinux at dosages suggested for prophylaxis of VTE is intensely recommended in all SARS-CoV-2-hospitalized patients; subjects with anticoagulant contraindications should be cured with limb compression. Prophylactic heparin treatment should be performed for all the hospitalization time and for 7–14 days after hospital discharge in presence of VTE risk factors. The employ of intermediate-dose LMWH could be used on an individual basis in subjects with numerous risk factors for VTE, while the employ of therapeutic doses of LMWH is at present not validated by sufficient evidence and cannot be suggested as a standard therapy for all SARS-CoV-2 subjects [131].
Although the violence of the pandemic may suggest the use of heroic treatments to reduce the frightening mortality that accompanies SARS-CoV-2 infection, we believe that experimental treatments should only be used within approved and controlled experimental protocols, the only ones that can provide useful and specify information on the validity of the treatments.
Data availability
Not applicable.
Code availability
Not applicable.
References
Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, Ren R, Leung KSM, Lau EHY, Wong JY, Xing X, Xiang N, Wu Y, Li C, Chen Q, Li D, Liu T, Zhao J, Liu M, Tu W, Chen C, Jin L, Yang R, Wang Q, Zhou S, Wang R, Liu H, Luo Y, Liu Y, Shao G, Li H, Tao Z, Yang Y, Deng Z, Liu B, Ma Z, Zhang Y, Shi G, Lam TTY, Wu JT, Gao GF, Cowling BJ, Yang B, Leung GM, Feng Z (2020) Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med 382(13):1199–1207
Chan JF-W, Yuan S, Kok K-H, To KK-W, Chu H, Yang J, Xing F, Liu J, Yip CC-Y, Poon RW-S, Tsoi H-W, Lo SK-F, Chan K-H, Poon VK-M, Chan W-M, Ip JD, Cai J-P, Cheng VC-C, Chen H, Hui CK-M, Yuen K-Y (2020) A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 395(10223):514–523
Driggin E, Madhavan MV, Bikdeli B, Chuich T, Laracy J, Bondi-Zoccai G, Brown TS, Nigoghossian CD, Zidar DA, Haythe J, Brodie D, Beckman JA, Kirtane AJ (2020) Cardiovascular considerations for patients, health care workers, and health systems during the coronavirus disease 2019 (COVID-19) pandemic. J Am Coll Cardiol 18 S0735-1097(20)34637-4
Bangash MN, Patel J, Parekh D (2020) COVID-19 and the liver: little cause for concern. Lancet Gastroenterol Hepatol 5(6):529–530. https://doi.org/10.1016/S2468-1253(20)30084-4
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ (2020) HLH across speciality collaboration, UK. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395(10229):1033–1034
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, Xia J, Yu T, Zhang X, Zhang L (2020) Epidemiological and clinical characteristics of 99 cases of novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395(10223):507–513. 2019
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 395(10223):497–506
Snijders D, Schoorl M, Schoorl M, Bartels PC, van der Werf TS, Boersma WG (2012) D-dimer levels in assessing severity and clinical outcome in patients with community-acquired pneumonia. A secondary analysis of a randomised clinical trial. Eur J Intern Med 23(5):436–441
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y, Zhao Y, Li Y, Wang X, Peng Z (2020) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA:e201585. https://doi.org/10.1001/jama.2020.1585
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, Guan L, Wei Y, Li H, Wu X, Xu J, Tu S, Zhang Y, Chen H, Cao B (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395(10229):1054–1062
Tang N, Li D, Wang X, Sun Z (2020) Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 18(4):844–847
Han H, Yang L, Liu R, Liu F, Wu K-L, Li J, Liu X-H, Zhu C-L (2020) Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med. https://doi.org/10.1515/cclm-2020-0188
Kitchens CS (2009) Thrombocytopenia and thrombosis in disseminated intravascular coagulation (DIC). Hematology/the Education Program of the American Society of Hematology 240–246.
Gralinski LE, Bankhead A 3rd, Jeng S, Menachery VD, Proll S, Belisle SE, Matzke M, Webb-Robertson BJ, Luna ML, Shukla AK, Ferris MT, Bolles M, Chang J, Aicher L, Waters KM, Smith RD, Metz TO, Law GL, Katze MG, McWeeney S, Baric RS (2013) Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. mBio 4(4)
Berri F, Rimmelzwaan GF, Hanss M, Albina E, Foucault-Grunenwald ML, Lê VB, Vogelzang-van Trierum SE, Gil P, Camerer E, Martinez D, Lina B, Lijnen R, Carmeliet P, Riteau B (2013) Plasminogen controls inflammation and pathogenesis of influenza virus infections via fibrinolysis. PLoS Pathog 9(3):e1003229
Panigada M, Bottino N, Tagliabue P, Grasselli G, Novembrino C, Chantarangkul V, Pesenti A, Peyvandi F, Tripodi A (2020) Hypercoagulability of COVID-19 patients in Intensive Care Unit. A report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost. https://doi.org/10.1111/jth.14850
Zhang C, Shi L, Wang F (2020) Liver injury in COVID-19: management and challenges. Lancet Gastroenterol Hepatol 5(5):428–430
Madjid M, Safavi-Naeini P, Solomon SD, Vardeny O (2020) Potential effects of coronaviruses on the cardiovascular system: a review. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2020.1286
Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, Gong W, Liu X, Liang J, Zhao O, Huang H, Yang B, Huang C (2020) Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol:e200950. https://doi.org/10.1001/jamacardio.2020.0950
Helms J, Tacquard C, Severac F et al (2020) CRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis). High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med:1–10. https://doi.org/10.1007/s00134-020-06062-x
Oxley TJ, Mocco J, Majidi S et al (2020) Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med 382(20):e60
Bangalore S, Sharma A, Slotwiner A et al (2020) ST segment elevation in patients with Covid-19 - a case series. N Engl J Med. https://doi.org/10.1056/NEJMc2009020
Bellosta R, Luzzani L, Natalini G et al (2020) Acute limb ischemia in patients with COVID-19 pneumonia. J Vasc Surg. https://doi.org/10.1016/j.jvs.2020.04.483
Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, Endeman H (2020) Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. https://doi.org/10.1016/j.thromres.2020.04.013
Lim W, Meade M, Lauzier F, Zarychanski R, Mehta S, Lamontagne F, Dodek P, McIntyre L, Hall R, Heels-Ansdell D, Fowler R, Pai M, Guyatt G, Crowther MA, Warkentin TE, Devereaux PJ, Walter SD, Muscedere J, Herridge M, Turgeon AF, Geerts W, Finfer S, Jacka M, Berwanger O, Ostermann M, Qushmaq I, Friedrich JO, Cook DJ, PROphylaxis for ThromboEmbolism in Critical Care Trial Investigators (2015) Failure of anticoagulant thromboprophylaxis: risk factors in medical-surgical critically ill patients multicenter study; randomized controlled trial. Crit Care Med 43(2):401–410
Cui S, Chen S, Li X, Liu S, Wang F (2020) Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. https://doi.org/10.1111/jth.14830
Zakai NA, McClure LA (2011) Racial differences in venous thromboembolism. J Thromb Haemost 9:1877–1882
Boisrame-Helms J, Kremer H, Schini-Kerth V, Meziani F (2013) Endothelial dysfunction in sepsis. Curr Vasc Pharmacol 11:150–160
Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT (2009) Extracellular histones are major mediators of death in sepsis. Nat Med 15(11):1318–1321
Boccia M, Aronne L, Celia B, Mazzeo G, Ceparano M, D’Agnano V, Parrella R, Valente Y, Bianco A, Perrotta F (2020) COVID-19 and coagulative axis: review of emerging aspects in a novel disease. Monaldi Arch Chest Dis 90(2). https://doi.org/10.4081/monaldi.2020.1300 Published 2020 May 19
Bucciarelli P, Martinelli I, Artoni A, Passamonti SM, Previtali E, Merati G, Tripodi A, Mannucci PM (2012) Circulating microparticles and risk of venous thromboembolism. Thromb Res 129:591–597
Raeven P, Zipperle J, Drechsler S (2018) Extracellular vesicles as markers and mediators in sepsis. Theranostics 8:3348–3365
Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, Nácher M, Pitaval C, Radovanovic I, Fukui Y, McEver RP, Filippi MD, Lizasoain I, Ruiz-Cabello J, Zarbock A, Moro MA, Hidalgo A (2014) Neutrophils scan for activated platelets to initiate inflammation. Science. 346:1234–1238
Bowles L, Platton S, Yartey N et al (2020) Lupus anticoagulant and abnormal coagulation tests in patients with COVID-19. N Engl J Med. https://doi.org/10.1056/NEJMc2013656
Tagami T, Tosa R, Omura M, Fukushima H, Kaneko T, Endo T, Rinka H, Murai A, Yamaguchi J, Yoshikawa K, Saito N, Uzu H, Kase Y, Takatori M, Izumino H, Nakamura T, Seo R, Kitazawa Y, Sugita M, Takahashi H, Kuroki Y, Irahara T, Kanemura T, Yokota H, Kushimoto S (2014) Effect of a selective neutrophil elastase inhibitor on mortality and ventilator-free days in patients with increased extravascular lung water: a post hoc analysis of the PiCCO Pulmonary Edema Study. J Intensive Care 2:67
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science. 303:1532–1535
Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, Menninge S, Eickhoff J, Nussbaumer P, Klebl B, Krüger R, Herzig A, Zychlinsky A (2018) Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol 3:eaar6689
Schonrich G, Raftery MJ (2016) Neutrophil extracellular traps go viral. Front Immunol 7:366
Papayannopoulos V (2018) Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18:134–147
Kapoor S, Opneja A, Nayak L (2018) The role of neutrophils in thrombosis. Thromb Res 170:87–96
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 107:15880–15,885
Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT (2011) Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb Haemost 9:1795–1803
Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, Gallant M, Martinod K, Cate HT, Hofstra L, Crijns HJ, Wagner DD, Kietselaer BLJH (2013) Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol 33:2032–2040
Laridan E, Martinod K, De Meyer SF (2019) Neutrophil extracellular traps in arterial and venous thrombosis. Semin Thromb Hemost 45:86–93
Jiménez-Alcázar M, Rangaswamy C, Panda R, Bitterling J, Yashin J, Long AT, Bilyy R, Krenn V, Renné C, Renné T, Kluge S, Panzer U, Mizuta R, Mannherz HG, Kitamura D, Herrmann M, Napirei M, Fuchs TA (2017) Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science. 358:1202–1206
Oehmcke S, Morgelin M, Herwald H (2009) Activation of the human contact system on neutrophil extracellular traps. J Innate Immun 1:225–230
Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT (2011) Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 118:1952–1961
Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, Konrad I, Kennerknecht E, Reges K, Holdenrieder S, Braun S, Reinhardt C, Spannagl M, Preissner KT, Engelmann B (2010) Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 16:887–896
Raup-Konsavage WM, Wang Y, Wang WW, Feliers D, Ruan H, Reeves WB (2018) Neutrophil peptidyl arginine deiminase-4 has a pivotal role in ischemia/reperfusion-induced acute kidney injury. Kidney Int 93:365–374
Campbell CM, Kahwash R (2020) Will complement inhibition be the new target in treating COVID-19 related systemic thrombosis? Circulation. https://doi.org/10.1161/CIRCULATIONAHA.120.047419
Lippi G, Plebani M, Henry BM (2020) Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta 506:145–148
Qu R, Ling Y, Zhang YH, Wei LY, Chen X, Li XM, Liu XY, Liu HM, Guo Z, Ren H, Wang O (2020) Platelet-to-lymphocyte ratio is associated with prognosis in patients with coronavirus disease-19. J Med Virol. https://doi.org/10.1002/jmv.25767
Yin S, Huang M, Li D, Tang N (2020) Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J Thromb Thrombolysis. https://doi.org/10.1007/s11239-020-02105-8
Menter DG, Kopetz S, Hawk E, Sood AK, Loree JM, Gresele P, Honn KV (2017) Platelet “first responders” in wound response, cancer, and metastasis. Cancer Metastasis Rev 36(2):199–213
Witkowski M, Landmesser U, Rauch U (2016) Tissue factor as a link between inflammation and coagulation. Trends Cardiovasc Med 26:297–303
Levi M, van der Poll T (2017) Coagulation and sepsis. Thromb Res 149:38–44
Schmitt FCF, Manolov V, Morgenstern J, Fleming T, Heitmeier S, Uhle F, Al-Saeedi M, Hackert T, Bruckner T, Schöchl H, Weigand MA, Hofer S, Brenner T (2019) Acute fibrinolysis shutdown occurs early in septic shock and is associated with increased morbidity and mortality: results of an observational pilot study. Ann Intensive Care 9(1):19
Wright FL, Vogler TO, Moore EE, Moore HB, Wohlauer MV, Urban S, Nydam TL, Moore PK, McIntyre RC Jr (2020) Fibrinolysis shutdown correlates to thromboembolic events in severe COVID-19 infection. J Am Coll Surg S1072–7515(20):30400–30402. https://doi.org/10.1016/j.jamcollsurg.2020.05.007
Terpos E, Ntanasis-Stathopoulos I, Elalamy I, Kastritis E, Sergentanis TN, Politou M, Psaltopoulou T, Gerotziafas G, Dimopoulos MA (2020) Hematological findings and complications of COVID-19. Am J Hematol. https://doi.org/10.1002/ajh.25829
Isidori AM, Minnetti M, Sbardella E, Graziadio C, Grossman AB (2015) Mechanisms in endocrinology: the spectrum of haemostatic abnormalities in glucocorticoid excess and defect. Eur J Endocrinol 173(3):R101–R113
Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, Wang Z, Li J, Li J, Feng C, Zhang Z, Wang L, Peng L, Chen L, Qin Y, Zhao D, Tan S, Yin L, Xu J, Zhou C, Jiang C, Liu L (2020) Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci 63:364–374
Verdecchia P, Cavallini C, Spanevello A, Angeli F (2020) The pivotal link between ACE-2 deficiency and SARS-CoV-2 infection. Eur J Intern Med:1–7. https://doi.org/10.1016/j.ejim.2020.04.037
Lin CI, Tsai CH, Sun YL, Hsieh WY, Lin YC, Chen CY, Lin CS (2018) Instillation of particulate matter 2.5 induced acute lung injury and attenuated the injury recovery in ACE-2 knockout mice. Int J Biol Sci 14:253–265
Bikdeli B, Madhavan MV, Gupta A, Jimenez D, Burton JR, Nigoghossian CD, Chuich T, Nouri SN, Dreyfus I, Driggin E, Sethi S, Sehgal K, Chatterjee S, Ageno W, Madjid M, Guo Y, Tang LV, Hu Y, Bertoletti L, Giri J, Cushman M, Quéré B, Dimakakos EP, Gibson CM, Lippi G, Favaloro EJ, Fareed J, Tafur AJ, Francese DP, Batra J, Falanga A, Clerkin KJ, Uriel N, Kirtane A, McLintock C, Hunt BJ, Spyropoulos AC, Barnes GD, Eikelboom JW, Weinberg I, Schulman S, Carrier M, Piazza G, Beckman JA, Leon MB, Stone GW, Rosenkranz S, Goldhaber SZ, Parikh SA, Monreal M, Krumholz HM, Konstantinides SV, Weitz JI, Lip GYHGlobal COVID-19 Thrombosis Collaborative Group (2020) Pharmacological agents targeting thrombo-inflammation in COVID-19: review and implications for future research. Thromb Haemost. https://doi.org/10.1055/s-0040-1,713,152
Gupta N, Zhao YY, Evans CE (2019) The stimulation of thrombosis by hypoxia. Thromb Res 181:77–83
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE-2 receptor. Nature. https://doi.org/10.1038/s41586-020-2180-5
Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A, Li F (2020) Structural basis of receptor recognition by SARS-CoV-2. Nature. https://doi.org/10.1038/s41586-020-2179-y
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E (2020) The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir Res 176:104742
Liu PP, Blet A, Smyth D, Li H (2020) The science underlying COVID-19: implications for the cardiovascular system. Circulation. https://doi.org/10.1161/CIRCULATIONAHA.120.047549
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. https://doi.org/10.1016/j.cell.2020.02.058
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Muller MA, Drosten C, Pohlmann S (2020) SARS-CoV-2 cell entry depends on ACE-2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. https://doi.org/10.1016/j.cell.2020.02.052
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454
Oudit GY, Kassiri Z, Jiang C, Liu PP, Poutanen SM, Penninger JM, Butany J (2009) SARS coronavirus modulation of myocardial ACE-2 expression and inflammation in patients with SARS. Eur J Clin Investig 39:618–625
Fung SY, Yuen KS, Ye ZW, Chan CP, Jin DY (2020) A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg Microbes Infect 9(1):558–570
Schultz MJ, Haitsma JJ, Zhang H, Slutsky AS (2006) Pulmonary coagulopathy as a new target in therapeutic studies of acute lung injury or pneumonia—a review. Crit Care Med 34(3):871–877
Kerr R, Stirling D, Ludlam CA (2001) Interleukin 6 and haemostasis. Br J Haematol 115:3–12
Bode M, Mackman N (2014) Regulation of tissue factor gene expression in monocytes and endothelial cells: thromboxane A2 as a new player. Vasc Pharmacol 62:57–62
Dolhnikoff M, Duarte-Neto AN, de Almeida Monteiro RA, Ferraz da Silva LF, de Oliveira EP, Saldiva PHN, Mauad T, Negri EM (2020) Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J Thromb Haemost. https://doi.org/10.1111/jth.14844
Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, Baxter-Stoltzfus A, Laurence J (2020) Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. https://doi.org/10.1016/j.trsl.2020.04.007
Prufer F, Scheiring J, Sautter S, Jensen DB, Treichl R, Würzner R, Zimmerhackl LB (2006) Terminal complement complex (C5b-9) in children with recurrent hemolytic uremic syndrome. Semin Thromb Hemost 32:121–127
Chaturvedi S, Braunstein EM, Yuan X, Yu J, Alexander A, Chen H, Gavriilaki E, Alluri R, Streiff MB, Petri M, Crowther MA, McCrae KR, Brodsky RA (2020) Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood 135:239–251
Zhou Y, Lu K, Pfefferle S, Bertram S, Glowacka I, Drosten C, Pöhlmann S, Simmons G (2010) A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J Virol 84:8753–8764
Beltrame MH, Catarino SJ, Goeldner I, Boldt ABW, de Messias-Reason IJ (2015) The lectin pathway of complement and rheumatic heart disease. Front Pediatr 2(148):1–14
Krarup A, Wallis R, Presanis JS, Gal P, Sim RB (2007) Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS One 7:e623
Campbell CM, Cassol C, Cataland SR, Kahwash R (2020) Atypical haemolytic uraemic syndrome: a case report of a rare cause of reversible cardiomyopathy. Eur Heart J - Case Rep. https://doi.org/10.1093/ehjcr/ytaa050
Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, Whitmore A, Heise MT, Baric RS (2018) Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio:9. https://doi.org/10.1128/mBio.01753-18
Jiang Y, Zhao G, Song N, Li P, Chen Y, Guo Y, Li J, Du L, Jiang S, Guo R, Sun S, Zhou Y (2018) Blockade of the C5a-C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg Microbes Infect 7:77
Rits K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, Rafail S, Kartalis G, Sideras P, Lambris JD (2006) A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol 177:47944802
Yen Y-T, Liao F, Hsiao C-H, Kao C-L, Chen Y-C, Wu-Hsieh BA (2006) Modeling the early events of severe acute respiratory syndrome coronavirus infection in vitro. J Virol 80:26842693
Fan BE, Chong VCL, Chan SSW, Lim GH, Lim KGE, Tan GB, Mucheli SS, Kuperan P, Ong KH (2020) Hematologic parameters in patients with COVID-19 infection. Am J Hematol. https://doi.org/10.1002/ajh.25774
Bhatraju PK, Ghassemieh BJ, Nichols M, Kim R, Jerome KR, Nalla AK, Greninger AL, Pipavath S, Wurfel MM, Evans L, Kritek PA, West TE, Luks A, Gerbino A, Dale CR, Goldman JD, O’Mahony S, Mikacenic C (2020) Covid-19 in critically ill patients in the Seattle region--case series. N Engl J Med. https://doi.org/10.1056/NEJMoa2004500
Peiro C, Moncada S (2020) Substituting angiotensin-(1–7) to prevent lung damage in SARSCoV2 infection? Circulation. https://doi.org/10.1161/CIRCULATIONAHA.120.047297
Lei C, Fu W, Qian K, Li T, Zhang D, Ding MX (2020) Potent neutralization of 2019 novel coronavirus by recombinant ACE-2-ig. bioRxiv. https://doi.org/10.1101/2020.02.01.929976
Verdecchia P, Cavallini C, Spanevello A, Angeli F (2020) COVID-19: ACE-2centric infective disease? Hypertension. https://doi.org/10.1161/HYPERTENSIONAHA.120.15353
Thachil J, Tang N, Gando S, Falanga A, Cattaneo M, Levi M, Clark C, Iba T (2020) ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost. https://doi.org/10.1111/JTH.14810
Poterucha TJ, Libby P, Goldhaber SZ (2017) More than an anticoagulant: Do heparins have direct anti-inflammatory effects? Thromb Haemost 117(3):437–444
Young E (2018) The anti-inflammatory effects of heparin and related compounds. Thromb Res 122(6):743–752
Li JP, Vlodavsky I (2009) Heparin, heparan sulfate and heparanase in inflammatory reactions. Thromb Haemost 102(5):823–828
Esmon CT (2014) Targeting factor Xa and thrombin: impact on coagulation and beyond. Thromb Haemost 111(4):625–633
Mousavi S, Moradi M, Khorshidahmad T, Motamedi M (2015) Anti-inflammatory effects of heparin and its derivatives: a systematic review. Adv Pharmacol Sci 2015:507151
Iba T, Hashiguchi N, Nagaoka I, Tabe Y, Kadota K, Sato K (2015) Heparins attenuated histone-mediated cytotoxicity in vitro and improved the survival in a rat model of histone-induced organ dysfunction. Intensive Care Med Exp 3(1):36
Zhu C, Liang Y, Li X, Chen N, Ma X (2019) Unfractionated heparin attenuates histone-mediated cytotoxicity in vitro and prevents intestinal microcirculatory dysfunction in histone-infused rats. J Trauma Acute Care Surg 87(3):614–622
Liu Y, Mu S, Li X, Liang Y, Wang L, Ma X (2019) Unfractionated heparin alleviates sepsis-induced acute lung injury by protecting tight junctions. J Surg Res 238:175–185
Ma J, Bai J (2015) Protective effects of heparin on endothelial cells in sepsis. Int J Clin Exp Med 8(4):5547–5552
Sun H, Wang X, Degen JL, Ginsburg D (2009) Reduced thrombin generation increases host susceptibility to group A streptococcal infection. Blood. 113(6):1358–1364
Hardaway RM, Williams CH, Marvasti M, Farias M, Tseng A, Pinon I, Yanez D, Martinez M, Navar J (1990) Prevention of adult respiratory distress syndrome with plasminogen activator in pigs. Crit Care Med 18:1413–1418
Konstantinides S, Geibel A, Heusel G, Heinrich F, Kasper W (2002) Management strategies and prognosis of pulmonary embolism-3 trial investigators. Heparin plus alteplase compared with heparin alone in patients with submassive pulmonary embolism. N Engl J Med 347:1143–1150
Yost CC, Schwertz H, Cody MJ, Wallace JA, Campbell RA, Vieira-deAbreu A, Araujo CV, Schubert S, Harris ES, Rowley JW, Rondina MT, Fulcher JM, Koening CL, Weyrich AS, Zimmerman GA (2016) Neonatal NET-inhibitory factor and related peptides inhibit neutrophil extracellular trap formation. J Clin Invest 126:3783–3379
Fuchs TA, Jimenez-Alcazar M, Gobel J, Englert H (2019) Neutrolis, Inc. US patent application US 2020/0024585 A1, filed August 20
Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR (2018) Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 3:e98178
Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, Daßler-Plenker J, Guerci P, Huynh C, Knight JS, Loda M, Looney MR, McAllister F, Rayes R, Renaud S, Rousseau S, Salvatore S, Schwartz RE, Spicer JD, Yost CC, Weber A, Zuo Y, Egeblad M (2020) Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med 217(6):e20200652. https://doi.org/10.1084/jem.20200652
Döring Y, Libby P, Soehnlein O (2020) Neutrophil extracellular traps participate in cardiovascular diseases: recent experimental and clinical insights. Circ Res 126(9):1228–1241
Lillicrap D (2020) Disseminated intravascular coagulation in patients with 2019-nCoV pneumonia. J Thromb Haemost 18(4):786–787. https://doi.org/10.1111/jth.14781
Iba T, Nisio MD, Levy JH, Kitamura N, Thachil J (2017) New criteria for sepsis-induced coagulopathy (SIC) following the revised sepsis definition: a retrospective analysis of a nationwide survey. BMJ Open 7(9):e017046. https://doi.org/10.1136/bmjopen-2017-017046
Ranucci M, Ballotta A, Di Dedda U, Bayshnikova E, Dei Poli M, Resta M, Falco M, Albano G, Menicanti L (2020) The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J Thromb Haemost. https://doi.org/10.1111/jth.14854
Stein PD, Kayali F, Olson RE, Milford CE (2004) Pulmonary thromboembolism in Asians/Pacific Islanders in the United States: analysis of data from the National Hospital Discharge Survey and the United States Bureau of the Census. Am J Med 116(7):435–442
Eikelboom JW, Hirsh J (2006) Monitoring unfractionated heparin with the aPTT: time for a fresh look. Thromb Haemost 96(05):547–552
Vandiver JW, Vondracek TG (2012) Antifactor Xa levels versus activated partial thromboplastin time for monitoring unfractionated heparin. Pharmacotherapy 32(06):546–558
Aikawa N, Shimazaki S, Yamamoto Y, Saito H, Maruyama I, Ohno R, Hirayama A, Aoki Y, Aoki N (2011) Thrombomodulin alfa in the treatment of infectious patients complicated by disseminated intravascular coagulation: subanalysis from the phase 3 trial. Shock 35(4):349–354
Liu XL, Wang XZ, Liu XX, Hao D, Jaladat Y, Lu F, Sun T, Lv CJ (2014) Low-dose heparin as treatment for early disseminated intravascular coagulation during sepsis: a prospective clinical study. Exp Ter Med 7(3):604–608
Nishida O, Ogura H, Egi M, Fujishima S, Hayashi Y, Iba T, Imaizumi H, Inoue S, Kakihana Y, Kotani J, Kushimoto S, Masuda Y, Matsuda N, Matsushima A, Nakada TA, Nakagawa S, Nunomiya S, Sadahiro T, Shime N, Yatabe T, Hara Y, Hayashida K, Kondo Y, Sumi Y, Yasuda H, Aoyama K, Azuhata T, Doi K, Doi M, Fujimura N, Fuke R, Fukuda T, Goto K, Hasegawa R, Hashimoto S, Hatakeyama J, Hayakawa M, Hifumi T, Higashibeppu N, Hirai K, Hirose T, Ide K, Kaizuka Y, Kan’o T, Kawasaki T, Kuroda H, Matsuda A, Matsumoto S, Nagae M, Onodera M, Ohnuma T, Oshima K, Saito N, Sakamoto S, Sakuraya M, Sasano M, Sato N, Sawamura A, Shimizu K, Shirai K, Takei T, Takeuchi M, Takimoto K, Taniguchi T, Tatsumi H, Tsuruta R, Yama N, Yamakawa K, Yamashita C, Yamashita K, Yoshida T, Tanaka H, Oda S (2018) The Japanese Clinical Practice Guidelines for Management of Sepsis and Septic Shock 2016 (J-SSCG 2016). Acure Med Surg 5(1):3–89
Umemura Y, Yamakawa K, Ogura H, Yuhara H, Fujimi S (2016) Efficacy and safety of anticoagulant therapy in three specific populations with sepsis: a meta-analysis of randomized controlled trials. J Thromb Haemost 14(3):518–530
Iba T, Gando S, Thachil J (2014) Anticoagulant therapy for sepsis-associated disseminated intravascular coagulation: the view from Japan. J Thromb Haemost 12(7):1010–1019
Arachchillage DRJ, Laffan M (2020) Correspondence on: abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. https://doi.org/10.1111/JTH.14820
Arachchillage DRJ, Passariello M, Laffan M, Aw TC, Owen L, Banya W, Trimlett R, Morgan C, Patel BV, Pepper J, Ledot S (2018) Intracranial hemorrhage and early mortality in patients receiving extracorporeal membrane oxygenation for severe respiratory failure. Semin Thromb Hemost 44(3):276–286
Li Z, Li X, Huang Y-Y, Zhou L, Liu R, Wu D, Zhang L, Liu H, Xu X, Zhang Y, Cui J, Wang X, Luo H-B (2020) FEP-based screening prompts drug repositioning against COVID-19. bioRxiv 2020 2020.03.23. 004580
Liu X, Li Z, Liu S, Sun J, Chen Z, Jiang M, Zhang Q, Wei Y, Wang X, Huang Y-Y, Shi Y, Xu Y, Xian H, Bai F, Ou C, Xiong B, Lew AM, Cui J, Fang R, Huang H, Zhao J, Hong X, Zhang Y, Zhou F, Luo H-B (2020) Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta PharmSin B. https://doi.org/10.1016/j.apsb.2020.04.008
Jose RJ, Manuel A (2020) COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med S2213–2600(20):30216–30212
Selvaraj V, Dapaah-Afriyie K, Finn A, Flanigan TP (2020) Short-term dexamethasone in Sars-CoV-2 Patients. R I Med J (2013) 103(6):39–43
Theoharides TC, Conti P (2020) Dexamethasone for COVID-19? Not so fast. J Biol Regul Homeost Agents 34(3). https://doi.org/10.23812/20-EDITORIAL_1-5
Marietta M, Ageno W, Artoni A, De Candia E, Gresele P, Marchetti M, Marcucci R, Tripodi A (2020) COVID-19 and haemostasis: a position paper from Italian Society on Thrombosis and Haemostasis (SISET). Blood Transfus. https://doi.org/10.2450/2020.0083-20
Author information
Authors and Affiliations
Contributions
Conceptualization, A A, C M; Data curation, V I, A G A; Writing (original draft), A A; Writing (review and editing) A A.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Allegra, A., Innao, V., Allegra, A.G. et al. Coagulopathy and thromboembolic events in patients with SARS-CoV-2 infection: pathogenesis and management strategies. Ann Hematol 99, 1953–1965 (2020). https://doi.org/10.1007/s00277-020-04182-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-020-04182-4