
Abstract
Livestock farms are major reservoirs of antibiotic resistance genes (ARGs) that are discharged into the environment. However, the abundance, diversity, and transmission of ARGs in duck farms and its impact on surrounding environments remain to be further explored. Therefore, the characteristics of ARGs and their bacterial hosts from duck farms and surrounding environment were investigated by using metagenomic sequencing. Eighteen ARG types which consist of 823 subtypes were identified and the majority conferred resistance to multidrug, tetracyclines, aminoglycosides, chloramphenicols, MLS, and sulfonamides. The floR gene was the most abundant subtype, followed by sul1, tetM, sul2, and tetL. ARG abundance in fecal sample was significantly higher than soil and water sample. Our results also lead to a hypothesis that Shandong province have been the most contaminated by ARGs from duck farm compared with other four provinces. PcoA results showed that the composition of ARG subtypes in water and soil samples was similar, but there were significant differences between water and feces samples. However, the composition of ARG subtypes were similar between samples from five provinces. Bacterial hosts of ARG subtypes were taxonomically assigned to eight phyla that were dominated by the Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria. In addition, some human bacterial pathogens could be enriched in duck feces, including Enterococcus faecium, Acinetobacter baumannii, and Staphylococcus aureus, and even serve as the carrier of ARGs. The combined results indicate that a comprehensive overview of the diversity and abundance of ARGs, and strong association between ARGs and bacterial community shift proposed, and benefit effective measures to improve safety of antibiotics use in livestock and poultry farming.
Key points
• ARG distribution was widespread in the duck farms and surroundings environment
• ARG abundance on the duck farms was significantly higher than in soil and water
• Human bacterial pathogens may serve as the vectors for ARGs
Similar content being viewed by others


Introduction
Antibiotics are critical for the prevention and treatment of bacterial infections and the spread of antibiotic resistance undermines their effectiveness (Dai et al. 2022). Antimicrobial resistance is a growing and major global health concern due to the high rates of morbidity and mortality that it can cause an estimated 10 million deaths by 2050 if unchecked (O'Neill 2014; Rodríguez et al. 2021). The excessive and indiscriminate use of antibiotics in both human and veterinary medicine has significantly accelerated the widespread development of antibiotic resistance. As a result, antibiotic resistance genes (ARGs) are now ubiquitous in the human-animal-environment interface (Li et al. 2021). In recent years, a deeper appreciation of the “One Health” concept has emerged, recognizing the links between the health of humans, animals, and the environment. This has led to the recognition that livestock and poultry farms and their surrounding environments are crucial links in the dissemination of antibiotic resistant organisms (Hooban et al. 2021). An increasing number of studies have demonstrated that swine feedlots and abattoirs, wild boars, fish, broiler chickens, and dairy farms are recognized the reservoirs of ARGs (Bai et al. 2021; Dias Diana 2021; He et al. 2019; Jo et al. 2021; Munk et al. 2018). However, the spread and distribution characteristics of ARGs in duck farms have been largely ignored. Duck production has the potential to play a major role in the agricultural economy, and Asian countries alone contribute 84.2% of total duck meat produced in the world. China is the largest global producer and consumer of cultivated ducks (Wang et al. 2021b). Thus, it is necessary to assess the ARG profile and distribution of duck farms in China.
Considerable evidence suggested that ARGs and antibiotics are released from livestock and poultry farms into their surroundings that include water, soil and air (Zhang et al. 2020c). For example, the airborne ARGs can disperse from the animal houses to a distance of 10 km along the wind direction in chicken and dairy farms (Bai et al. 2021). ARGs are also released from stored swine manure biogas digestate to the atmosphere via aerosol dispersion (Zhang et al. 2020c). These bioaerosols play keys roles in transmission of antibiotic resistance, and levels of airborne ARGs from pig farms are elevated during the winter months (Song et al. 2021). In addition, swine farming can enhance the levels of veterinary antibiotics and ARGs in groundwater (Gao et al. 2020). Furthermore, a lack of sewage treatment techniques in intensive breeding has led to ARG contamination of neighboring fishponds and aquaculture water and associated ARGs are discharged directly into rivers, lakes and seas without treatment (Fu et al. 2022; He et al. 2019). After that, ARGs can also persist in aquatic environments and this promotes further dissemination of ARGs between aquatic environment and aquatic animals (Jo et al. 2021; Mahaney and Franklin 2022).
Composting is an economical and environmentally friendly approach to treat cattle, swine, and chicken manure (Wang et al. 2022; Xu et al. 2022). The elevated temperatures produced during composting can decrease ARG abundance for some bacterial taxa, and some ARGs have proven recalcitrant to this process (Mao et al. 2021). Agricultural products can also be contaminated with ARGs through manure or to a lesser extent by compost and consequently are a potential health risk to both animals and humans (He et al. 2019). ARG migration to deeper soil layers by long-term land application of animal manures has increased the complexity of the problem (Tang et al. 2015). Therefore, ARG monitoring on duck farms and their surrounding environment is the first step into assessing whether this poses a health threat.
Bacterial community shifts are key factors driving changes of ARG profiles in different host habitats (Guo et al. 2021). Generally, swine manure is a diverse bacterial source and the genera Romboutsia, Clostridisensu_stricto_1, and Terrisporobacter are the primary bacterial ARG hosts found during swine manure composting (Wang et al. 2021a). However, the Streptococcaceae are also sources of multidrug, MLS, and aminoglycoside ARGs in pig feces (Zhang et al. 2021), and Prevotellaceae and Ochrobactrum are enriched with tetracycline ARGs in airborne samples (Song et al. 2021). Metagenomic assembly–based host-tracking analysis identified Escherichia, Bacteroides, and Clostridium as the predominant bacterial hosts of ARGs in gut-associated environments and pristine environments (Zeng et al. 2019). Our previous work indicated a major presence of carbapenemase-producing Enterobacteriaceae on duck farms and their surrounding environments (Wang et al. 2021c). Therefore, this prompted us to explore the roles of bacterial community shifts in the resistome alterations that originate on duck farms and surrounding environment.
In this study, duck feces and surrounding environmental samples were collected from duck farms in south-east coastal China including Shandong, Jiangsu, Zhejiang, Fujian, and Guangdong province. The main objectives of the study are to compare the diversity and abundance of ARGs in duck feces and their surrounding environment, correlate ARGs with the bacterial community and further predict the potential ARGs host, and explore ARG-associated human pathogenic bacteria. These findings will contribute to the full understanding of ARGs, ARG-hosts, and ARG-associated human pathogenic bacteria in duck farms and their surrounding environment.
Materials and methods
Sample collection
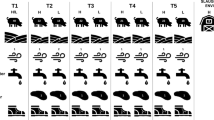
Feces and environmental samples were collected from 2018 to 2019 at 29 duck farms in Shandong, Jiangsu, Zhejiang, Fujian, and Guangdong, China, including 29 fecal, 32 soil, and 24 water samples (Fig. 1A). Fresh feces were placed in 10 mL sterile tubes. soil samples from each duck farm were collected using a five-point sampling method and were collected at depth 5 to 10 cm and transferred to sterile bags. Notably, the farms in Shandong province used an automatic water system and ducks are raised in cages with approximately 10 individuals per cage 1 m above the ground. In the other four provinces, ducks were fed and watered together houses of about 10,000 individuals and range freely in ponds and rivers (Fig. 1B). Therefore, the water samples in Jiangsu, Zhejiang, Fujian, and Guangdong Province were collected from uniformly selected sampling points near ponds and rivers in duck farms; water samples in Shandong Province are mainly collected in automatic water supply systems; and 500 mL water samples were collected from per sampling site and three to five sampling sites were selected per farm. All the farms were in use for at least 3 years and each produce > 10,000 commercial ducks annually. All samples were kept on dry ice for transportation to the laboratory and stored at − 80 °C before processing.

Schematic diagram of sample collection and farming patterns. A Samples were collected from 29 duck farms and surrounding environment in five southeastern coastal provinces of China. B Farming patterns in most of duck farms in southeastern coastal provinces of China
DNA extraction
Samples collected at the same sampling site were mixed for metagenomic DNA extraction. For feces and soil samples, metagenomic DNA was extracted from approximately 0.25 g feces or soil using the QIAamp Power Fecal DNA isolation kit and DNeasy PowerSoil kits (Cat No 12830–50; Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Water samples were filtered with sterile 0.45 μm filters, and metagenomic DNA was also extracted from the filter membranes using the DNeasy Power Water kit (Cat No 14900–50-NF, Qiagen). The quality, purity, and yield of the extracted DNA were quantified using 1% agarose gel electrophoresis and a NanoDrop2000 spectrophotometer (Thermo Fisher, Pittsburg, PA, USA), and Qubit 2.0 fluorometer (Thermo Fisher). The qualified DNA was stored at − 80 °C until further processing.
Metagenomic sequencing
Metagenomic libraries were generated from 1 μg DNA per sample using the TruSeq DNA Sample Prep Kit (Illumina, San Diego, CA, USA) following the manufacturer’s recommendations. Briefly, DNA samples were sheared ultrasonically to 250–300 base pair fragments and end-polished, A-tailed, and ligated with the dual index adaptors for Illumina sequencing with further PCR amplification. All libraries were then sequenced using an Illumina Hiseq 2500 platform with 2 × 150 bp paired reads (Novogene, Beijing, China). Raw reads were quality trimmed by using Trimmomatic version 0.32 to remove paired reads with adapter, paired reads should be removed when the number of low-quality (Q ≤ 5) bases in single-ended sequencing reads exceeds 50% of the read length ratio.
Analysis of ARGs
The relative abundance of ARGs was determined using the SARG 2.0 database (Yin et al. 2018). Putative ARG sequences were screened using USEARCH and normalized by the length. The sequence of extracted was annotated and classified using BLASTX with default parameters (the cutoff of e value of 10−7, sequence identity of 80%, and alignment length > 25 amino acids) (Feng et al. 2018). The relative abundance of ARGs was normalized with the ppm units (Zhang et al. 2020b).
Analysis of ARG-carrying bacteria
Short reads in each sample were de novo assembled into contigs using MEGAHIT v1.1.3 with the default k-mer size (Li et al. 2015b). Open reading frames (ORFs) from each sample were searched against the ARG-OAP to identify the potential ARG-like ORFs using the BLASTP algorithm with the following parameters: sequence identity ≥ 80%, alignment coverage ≥ 70%, and e-value ≤ 1e − 5 (Xiong et al. 2018). Contigs harboring ARG-like ORFs were extracted using Prodigal software with the “meta” model (Hyatt et al. 2010). The taxonomy of the ARG-carrying bacterial communities was determined by annotating ARG-carrying contigs using Contig Annotation Tool, and the total reads were annotated using kraken2 with default parameters (von Meijenfeldt et al. 2019; Wood et al. 2019). The taxonomic list of the taxa at the species level was matched with a bacterial pathogen database (Li et al. 2015a). The mapped pathogens and the corresponding abundances were extracted for analysis.
Statistical analysis and data visualization
ARG diversity analysis and principal coordinate analysis (PCoA) were measured using the “vegan” packages (Oksanen 2018). Venn diagrams were graphed by ImageGP (http://www.ehbio.com/ImageGP). The correlation analysis was diagrammed by “circlize” package (Gu et al. 2014). The heatmaps were visualized with the “pheatmap” package (Kolde 2019). The other figures were plotted by “ggplot2” and colored with “RColourBrewer” packages (Ginestet 2011; Neuwirth 2014).
Data availability
All sequence data of 82 samples were deposited NCBI Sequence Read Archive (Accession No. PRJNA848116).
Results
Overall view of ARG in duck farms and surroundings environment
A total of 82 metagenomic datasets were obtained from 85 samples collected from duck farms and surroundings environment, and DNA extraction failed from only thre samples. A total of 18 ARG types consisting of 823 subtypes from our 82 duck farms and surroundings environment samples. The six most dominant ARG types conferred resistance to multidrug (833 ± 1194 ppm) tetracyclines (574 ± 540 ppm), aminoglycosides (464 ± 507 ppm), chloramphenicols (323 ± 403 ppm), macrolide-lincosamide-streptogramins (MLS, 259 ± 290 ppm), and sulfonamides (209 ± 225 ppm). The sum of these six most dominant ARG types accounted for 58.4 ~ 94.7% of the total ARG relative abundance across the different samples (Fig. 2).

The rates of ARGs type prevalence across samples
Among these 823 subtypes, 529 subtypes encoded resistance to β-lactams followed by multidrug (n = 80), MLS (n = 48), aminoglycoside (n = 42), tetracycline (n = 44), and trimethoprim (n = 20) (Table S1). The 10 most abundant ARGs for each sample were extracted, and totally, 62 subtypes were shown in Fig. 3, with relative abundance levels of 56.54 ~ 94.16% of the corresponding total ARG relative abundance. The gene floR was the most abundant subtype encountered (6.91%, 159 ± 238 ppm) in all samples, followed by chloramphenicol exporters (5.49%, 126 ± 156 ppm), sul1 (5.14%, 118 ± 138 ppm), tetM (5.00%, 115 ± 133 ppm), sul2 (4.30%, 99 ± 104 ppm), and tetL (3.75%, 86 ± 139 ppm) (Table S2 and Table S3).

The 10 most abundant ARGs for each sample that totaled 62 subtypes for 82 metagenomic samples. In the main figure, each column represents one metagenome. Each row represents one ARG subtype. Abundance was transformed from the original abundance using the following formula: log10.ARG relative abundance (unit: ppm)
Comparison of ARG relative abundance among the different samples
At the type level, relative ARG abundance in fecal sample (6512 ± 2074 ppm) was significantly higher than soil and water samples (1202 ± 1152 ppm and 1217 ± 1226 ppm; P < 0.0001) (Fig. 4A). ARG relative abundance could also be ranked by province as from high to low were Shandong (4702 ± 1267 ppm), Jiangsu (3700 ± 3166 ppm), Fujian (3282 ± 2699 ppm), Guangdong (3199 ± 3755 ppm), and Zhejiang (2042 ± 2405 ppm) (Fig. 4B). There was no significant difference for ARG relative abundance between soil and fecal samples in Shandong (P > 0.05), while ARG relative abundance in fecal is significantly higher than soil in other provinces (P < 0.001) (Fig. 4C). These data indicated that duck feces are the reservoirs of ARGs.

Comparison of total ARG relative abundance across samples. A Sample type, B province, and C between origin in the same province. Boxes denote the interquartile (IQR) between the first and third quartiles (25th and 75th percentiles, respectively) and the line inside denotes the median. Whiskers denote the lowest and highest values within 1.5 times and the IQR from the first and third quartiles, respectively. (ns, P > 0.05; ***, P < 0.001; ****, P < 0.0001)
We identified six dominant ARG types in our sample collection, and multidrug, tetracycline, aminoglycoside, chloramphenicol, MLS, and sulfonamide ARGs were more abundant in fecal samples compared with soil and water samples. In the latter, the dominant ARG types were significantly higher in soil compared with water (P < 0.05), except multidrug (Table S4). In addition, the relative abundance of tetracycline, aminoglycoside, chloramphenicol, MLS, and sulfonamide resistance genes in all samples from Shandong were more abundant than other provinces. However, multidrug ARGs in all samples from Jiangsu and Guangdong were more abundant than Shandong, Zhjiang and Fujian (Table S5).
At the subtype level, the relative abundance of dominant ARG subtypes in fecal samples exceeded those in soil and water samples, although 6 subtypes (floR, chloramphenicol exporter, tetM, sul2, aadA, ermB) were enriched in soil samples compared with water. In contrast, sul1, multidrug transporter, aph(6)-I, aph(3″)-I, and tetA were lower in the soil samples (Table S2). The six dominant ARG subtypes (floR, chloramphenicol exporter, sul1, tetM and sul2) were more abundant in Shandong samples, while tetL in Fujian samples exceeded those from the other provinces (Table S3).
Comparison of ARG composition
A similarity analysis of ARG subtypes composition among 77 samples was performed using PCoA. In particular, the first two PCoA components of Bray–Curtis distance explained a high proportion of variance (57.98%) and the composition of ARGs subtypes were similar between water samples and soil samples and significantly different from that of the fecal samples. The ARG subtypes in feces were also closely clustered (Fig. 5A). Moreover, although the composition of ARGs subtypes were similar between samples from five provinces, the samples from Shandong were closely clustered and samples from Zhejiang, Fujian, and Guangdong possessed a level of variance (Fig. 5B).

Composition similarity and shared and unique ARG subtypes among different samples. ARG subtype compositions A for sample type and B by province. Numbers of shared and unique ARG subtypes by C sample type and D province
A calculation of shared and unique ARG subtypes indicated that 468/823 ARGs were shared by fecal, soil, and water samples. The fecal (n = 675) and water (n = 666) samples harbored exceeded that of the soil samples (n = 573) (Fig. 5C). In addition, the numbers of ARGs subtypes could be ranked as Guangdong (n = 665), Fujian (n = 655), Jiangsu (n = 619), Shandong (n = 545), and Zhejiang (n = 542) (Fig. 5D), indicating more diverse resistomes in samples from Guangdong, Fujian, and Jiangsu. In addition, the most abundant ARG types were multidrug resistance genes both in fecal (30.2%) and water samples (23.7%), while tetracycline resistance gene was the dominant ARG type in soil samples, accounting for 22.3% (Fig. S1).
ARG bacterial hosts
A total of 8567 ARG-carrying contigs were obtained from 77 samples through metagenomic assembly and aligned to the structured ARG database. Within this group, 25.7% (2205/8567) of the contigs were taxonomically assigned to eight phyla that were dominated by the Proteobacteria (1188/2205, 53.9%), Firmicutes (756/2205, 34.3%), Bacteroidetes (136/2205, 6.2%), and Actinobacteria (118/2205, 5.4%). The dominant bacterial phyla were Proteobacteria and Firmicutes in all of the samples, while Actinobacteria were higher than Bacteroidetes in the soil and water samples (Table S6).
In addition, only 9.9% (847/8567) contigs were classified into 41 bacterial families that were primarily composed of Moraxellaceae (228/847, 34.0%), Enterococcaceae (174/847, 20.5%), Enterobacteriaceae (63/847, 7.4%), Comamonadaceae (49/847, 5.8%), and Pseudomonadaceae (34/847, 4.0%) (Fig. S2 and Table S7). Notably, vancomycin ARGs were primarily harbored by Enterococcaceae (Fig. S2A). In contrast, the bacA resistance gene possessed the greatest host diversity (Fig. S2B). Fecal, soil, and water samples possessed 496, 158, and 193 contigs that could be aligned into the 27, 28, and 26 ARG-carrying bacterial families, respectively, and 16 families were shared across the 3 sample types (Fig. S3A). The numbers of ARG-carrying bacterial families were as follows 29 from Fujian, 25 from Shandong, 22 from Zhejiang, 20 from Guangdong, and 28 from Zhejiang, and included eight presented in all provinces (Fig. S3B). These distinctions were further defined and Moraxellaceae predominated in all three sample types, while Comamonadaceae levels in water exceeded that of soil and fecal samples and the Pseudomonadaceae equally represented in water samples and soil samples. In addition, the proportion of ARGs carried by Moraxellaceae was highest in Shandong, Jiangsu, and Zhejiang samples, while ARGs carried by Enterococcaceae were highest Fujian and Guangdong samples (Fig. 6A). ARGs distributed in Staphylococcaceae to a lesser degree although a higher percentage was detected in samples from Shandong (Fig. S4).

Comparisons of ARG bacterial hosts in feces, soil, and water. A ARG composition by sample type. B Similarity of ARG-carrying host compositions among sample types. C Similarity of ARG-carrying host compositions for Shandong, Jiangsu, Zhejiang, Fujian, and Guangdong
A similarity analysis of ARG-carrying bacterial family composition among 77 samples was also conducted, and PCoA revealed that the composition of ARG-carrying bacterial families in soil was similar to fecal and water samples, although fecal and water samples differed significantly (Fig. 6B). Furthermore, the composition of ARG-carrying bacterial families in samples from Shandong was closely clustered, whereas samples from Zhejiang, Fujian, and Guangdong exhibited evident scattering (Fig. 6C).
Characterization of ARG-carrying HPB
In this study, only 2.5% (212/8567) of the contigs could be classified and these included 59 bacterial species. Of which, the highest proportions were found for Enterococcus cecorum (74/212, 34.91%), Enterococcus faecium (14/212, 6.6%), Erysipelotrichaceae bacterium MTC7 (11/212, 5.2%), Acinetobacter baumannii (9/212, 4.2%), and Acinetobacter calcoaceticus (9/212, 4.2%) (Table S8). A total of 10 ARG-carrying human pathogenic bacteria (HPB) were detected in all samples though compared to bacterial pathogen database which contains 538 pathogenic species and distributed in 41 samples. In addition, Enterococcus faecium, Acinetobacter baumannii, and Staphylococcus aureus were detected in 14 (14/41, 34.1%), 9 (9/41, 22.0%), and 5 (5/41, 12.2%) samples, respectively. These numbers of ARGs-carrying HPB were twofold and fivefold higher in fecal compared with soil and water samples, respectively (Table S9).
Correlations between pathogenic bacteria, ARGs subtypes, and location of sample indicated that E. faecium were most likely to harbor the chloramphenicol resistance gene cat and distributed across multiple provinces. All A. baumannii carried the trimethoprim resistance gene dfrA20 and predominated in Jiangsu provinces. The Staphylococcus aureus mainly carried MLS resistance gene vgaE, and detected in Shandong and Fujian provinces. It is important to mention that Shandong province have been severely contaminated by a wide variety of ARGs-carrying HPB (Fig. S5).
Discussion
Distribution characteristics of ARGs
In this study, metagenomic analysis was used to detect the diversity and abundance of ARGs in duck farms and their surrounding environment. A total of 18 ARG types were identified in all the samples and dominated by multidrug, tetracycline, aminoglycoside, chloramphenicol, MLS, and sulfonamide resistant genes. These six were identical in ranking to those found on pig farms excepting that multidrug resistance genes were higher for duck farms aminoglycoside and sulfonamide resistance genes were lower than pig farms (Zhang et al. 2021). In addition, the relative abundance of ARGs in fecal samples was higher than for water and soil samples, which may pose a potential pollution threat to the surrounding environment. Meanwhile, ARGs have the potential to contaminate surrounding underground drinking water supplies via pig farm wastewater (He et al. 2016). The relative abundance of ARGs for pig farms were highest in Guangdong and Heilongjiang relative to Sichuan and Hebei (Wang et al. 2019). In contrast, we found that Guangdong levels were the lowest and Shandong the highest. In addition, ARG-carrying bacteria were highly endemic in backyard animals, commercial broiler farms, surrounding environment, and in the food chain in Shandong (Li et al. 2019; Wang et al. 2017). Collectively, these studies indicated that food animals in Shandong province may be serving as an enormous ARG reservoir. This indicates that farms are generally contaminated by ARGs and it is necessary to conduct continuous monitoring of ARGs in food-borne animals in Shandong Province and other provinces of China.
A total of 823 ARG subtypes were detected in this study, and among the 62 most prevalent ARG subtypes, floR, chloramphenicol exporter, sul1, tetM, sul2, and tetL were the most common. A total of 257 ARG subtypes had been previously detected in pig farms, and tet(W), tet(Q), tet(44), tet(37), and tet(40) were the predominant resistance gene (Joyce et al. 2019). These results suggest that the prevalence of sulfonamides and tetracycline resistance genes in duck and pig farms may be associated with the high frequency of use of these two drugs and sulfonamides and tetracycline drugs should be used reasonably in livestock and poultry breeding. Furthermore, ARG composition was more diverse in broiler chicken farms compared with the pig farms (Munk et al. 2018). In addition, predominant ARG subtypes in fecal samples were higher than environmental samples. PCoA indicated that composition of ARGs in soil and water were similar and only distantly related to the fecal samples. Therefore, the distribution of ARG subtypes may be primarily influenced by sample types (Mencia-Ares et al. 2020). It is also noteworthy that the relative abundance of floR was the highest in Zhejiang, Jiangsu, and Guangdong and the highest diversity was found in Guangdong and Fujian. These results were similar to prior studies on different pig farms in European countries that indicated a stark difference in diversity and relative abundance in different fecal samples (Munk et al. 2018). Overall, the diversity and relative abundance of ARGs may be influenced by geography.
Bacterial hosts
Bacterial communities are key factors shaping ARG prevalence. However, there is not a clear co-occurrence pattern of ARG subtypes and specific bacteria and the assembly of long ARG-carrying contigs is required for a more accurate taxonomic classification of ARGs (Zhang et al. 2019). Therefore, ARG-carrying contigs were extracted and Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria were identified as the major ARG hosts; the results in duck farms were similar to swine, chicken, and wild animals. For instance, Proteobacteria and Firmicutes predominated swine manure (Wang et al. 2021a; Zhang et al. 2021), while in dairy cow manure, ARGs originated from Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria (Wichmann et al. 2014). Proteobacteria was the predominant bacterial host and harbored the most diverse ARGs in broiler chickens and migratory birds (Cao et al. 2020).
We identified seven bacterial families (Moraxellaceae, Enterococcaceae, Enterobacteriaceae, Comamonadaceae, Pseudomonadaceae, Aeromonadaceae, and Staphylococcaceae as major potential ARG hosts. The Moraxellaceae hosted multidrug and bacitracin resistance genes including bacA, mexT, adeJ, adeK, and abaQ, although multidrug, MLS, and aminoglycoside resistance genes dominated for this family in swine manure (Zhang et al. 2021). Moraxellaceae were primarily represented by Acinetobacter that was the most prevalent ARG host (Zhang et al. 2020a). Acinetobacter were the predominated sulfonamide resistant bacteria in wastewater and shrimp ponds (Phuong Hoa et al. 2008). Multidrug resistance genes were carried by Enterobacteriaceae, and bacitracin resistance genes were carried by Enterococcaceae and Comamonadaceae. In the giant panda intestine, Enterobacteriaceae have been previously documented as the primary contributors to multidrug and tetracycline resistant genes and Enterococcaceae were associated with MLS carriage (Hu et al. 2020). The Burkholderiaceae were more prevalent in increasing multidrug resistance genes abundance in activated sludge reactors treated antibiotic production wastewater (Zhao et al. 2020). It is noteworthy that the multidrug resistance gene bacA possessed the greatest host diversity for the duck farms and surrounding environment. In contrast, Comamonadaceae were assigned as the major potential hosts of bacA in swine manure, wastewater, and soil (Zhang et al. 2021). In addition, the frequent hosts of bacA were Aeromonadaceae and Fusobacteria in a full-scale drinking water treatment plant (Jia et al. 2020). This is evidenced by our data which show that the potential bacterial hosts of ARGs are diverse, increasing the risk of transmission of ARGs between different bacterial species. In addition, this investigation also provided a scientific basis for taking measures to prevent the spread of resistance in farms.
ARG-carrying human pathogenic bacteria
Bacterial pathogens are the major concerns regarding public health, and we found ten ARG-carrying human pathogens, which included E. faecium, A. baumannii, and S. aureus that are members of the antimicrobial-resistant ESKAPE pathogens that also include E. faecium, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and Enterobacter spp (De Oliveira et al. 2020). These ESKAPE pathogens have reduced treatment options for serious infections and increased the burden of disease and death (De Oliveira et al. 2020). Our previous study obtained a small number of carbapenem-resistant K. pneumoniae and P. aeruginosa within the samples of the same batch (Wang et al. 2021c). Coincidentally, several zoonotic food-borne pathogens including Salmonella and Clostridium perfringens have also been detected on duck farms in China (Chen et al. 2020; Liu et al. 2022; Xiu et al. 2020). Thus, continued vigilance for ESKAPE pathogens in food animals is necessary especially since fecal serve as a reservoir for contamination of the surrounding environment (Yang et al. 2020).
Correlation analysis for human pathogenic bacteria and ARGs indicated that E. faecium was linked to chloramphenicol resistance genes, A. baumannii and S. aureus to sulfonamide and MLS resistance genes, respectively. E. faecium was also to harbor resistance to vancomycin (vanA, B, C, D, G, L, M, N, P), phenicol-oxazolidinone (poxtA), and linezolid (optrA) (Cattoir and Giard 2014). ARG analysis of A. baumannii genomes in public databases indicted that almost identical resistance genes for all isolates; armA, aph(3′)-VI-a, aph(6′)-Id, and strA genes all conferring resistance to aminoglycosides, tetB gene encoding for tetracycline resistance, mphE and mrsE genes, both responsible for macrolide resistance and sul1 and sul2 genes for sulphonamide resistance (Jia et al. 2019). Correspondingly, S. aureus harbored multiple ARGs collectively conferring resistance to aminoglycosides (aph(6)-Id, aph(3′)-III), β-lactam (blaZ, mecA), chloramphenicol (fexA), fosfomycin (fosB), lincosamide (lnuA), MLS (ermB, ermC), tetracyclines (tetL, tetM tetK), and trimethoprim (dfrA1, dfrG) (Gu et al. 2020). Importantly, the E. faecium and S. aureus harbored multiple ARGs that were widely distributed across retail samples of raw beef, sheep, and lamb meat, and were found in urban settings and can cause life-threatening blood stream infections due to limited treatment options (Gu et al. 2020; Pınar 2021; Wang et al. 2021d). Human pathogenic bacteria infections remain among the major worldwide causes of morbidity and mortality, even more worryingly, the emergence and transmission of ARG-carrying human pathogenic bacteria posed a growing global threat to human health. Therefore, to arrest the dissemination of resistant human pathogenic bacteria, these farms should develop more rational rules for antibiotic use.
This study has several limitations. First, this investigation neglected longitudinal monitoring to examine ARG persistence with their bacterial hosts. Second, antibiotic residue detection was lacking in this study. Therefore, the association between the antibiotic residues and ARG diversity requires further investigation. Third, environmental samples were not comprehensive enough and farm worker, drinking water, dust, flies, and aerosol samples were not collected. Therefore, the potential exposure risks of ARGs in duck farms still needs to be further studied.
In conclusion, a total of 18 ARG types consisting of 823 subtypes were detected in the duck farms and surrounding environment and the six dominant ARG types conferred resistance to multidrug, tetracycline, aminoglycoside, chloramphenicol, MLS, and sulfonamide. Among these 823 subtypes, floR was the most abundant in all samples followed by chloramphenicol exporter, sul1, tetM, sul2, and tetL. At the type level, the relative abundance of ARGs in feceal samples was significantly higher than soil and water samples. PCoA revealed that the composition of ARG subtypes were similar between water and soil samples and significantly difference from fecal samples. However, the composition of ARG subtypes were similar between samples from all five provinces. Bacterial ARG hosts were taxonomically assigned to eight phyla that were dominated by the Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria, and 41 bacterial families were deduced as the potential ARG hosts and Moraxellaceae, Enterococcaceae, Enterobacteriaceae, Comamonadaceae, and Pseudomonadaceae carried more diverse ARG subtypes than other families. In addition, some bacterial pathogens could be enriched in duck feces and could serve as ARG carriers. This study provides a comprehensive overview of the diversity and abundance of ARGs in duck farms and highlights the possible role of duck feces as ARG disseminators into the environment.
Data availability
All data generated or analyzed during this study are included in this article.
References
Bai H, He LY, Wu DL, Gao FZ, Zhang M, Zou HY, Yao MS, Ying GG (2021) Spread of airborne antibiotic resistance from animal farms to the environment: Dispersal pattern and exposure risk. Environ Int 158:106927. https://doi.org/10.1016/j.envint.2021.106927
Cao J, Hu Y, Liu F, Wang Y, Bi Y, Lv N, Li J, Zhu B, Gao GF (2020) Metagenomic analysis reveals the microbiome and resistome in migratory birds. Microbiome 8(1):26. https://doi.org/10.1186/s40168-019-0781-8
Cattoir V, Giard J-C (2014) Antibiotic resistance in Enterococcus faecium clinical isolates. Expert Rev Anti Infect Ther 12(2):239–248. https://doi.org/10.1586/14787210.2014.870886
Chen Z, Bai J, Wang S, Zhang X, Zhan Z, Shen H, Zhang H, Wen J, Gao Y, Liao M, Zhang J (2020) Prevalence, antimicrobial resistance, virulence genes and genetic diversity of Salmonella isolated from retail duck meat in Southern China. Microorganisms 8(3):444. https://doi.org/10.3390/microorganisms8030444
Dai D, Brown C, Bürgmann H, Larsson DGJ, Nambi I, Zhang T, Flach C-F, Pruden A, Vikesland PJ (2022) Long-read metagenomic sequencing reveals shifts in associations of antibiotic resistance genes with mobile genetic elements from sewage to activated sludge. Microbiome 10(1):20. https://doi.org/10.1186/s40168-021-01216-5
De Oliveira DMP, Forde BM, Kidd TJ, Harris PNA, Schembri MA, Beatson SA, Paterson DL, Walker MJ (2020) Antimicrobial resistance in ESKAPE pathogens. Clin Microbiol Rev 33(3):e00181-00119. https://doi.org/10.1128/CMR.00181-19
Dias Diana FC, S’onia M, Tˆania C (2021) A closer look on the variety and abundance of the faecal resistome of wild boar. Environ Pollut. https://doi.org/10.1016/j.envpol.2021.118406
Feng J, Li B, Jiang X, Yang Y, Wells GF, Zhang T, Li X (2018) Antibiotic resistome in a large-scale healthy human gut microbiota deciphered by metagenomic and network analyses. Environ Microbiol 20(1):355–368. https://doi.org/10.1111/1462-2920.14009
Fu C, Ding H, Zhang Q, Song Y, Wei Y, Wang Y, Wang B, Guo J, Qiao M (2022) Comparative analysis of antibiotic resistance genes on a pig farm and its neighboring fish ponds in a lakeside district. Environ Pollut 303:119180. https://doi.org/10.1016/j.envpol.2022.119180
Gao FZ, Zou HY, Wu DL, Chen S, He LY, Zhang M, Bai H, Ying GG (2020) Swine farming elevated the proliferation of Acinetobacter with the prevalence of antibiotic resistance genes in the groundwater. Environ Int 136:105484. https://doi.org/10.1016/j.envint.2020.105484
Ginestet C (2011) ggplot2: Elegant Graphics for Data Analysis. J R Stat Soc Ser A-Stat Soc 174:245–245. https://doi.org/10.1111/j.1467-985X.2010.00676_9.x
Gu ZG, Gu L, Eils R, SchlesnerBrors MB (2014) Circlize implements and enhances circular visualization in R. Bioinformatics 30(19):2811–2812. https://doi.org/10.1093/bioinformatics/btu393
Gu J, Xie XJ, Liu JX, Shui JR, Zhang HY, Feng GY, Liu XY, Li LC, Lan QW, Jin QH, Li R, Peng L, Lei CW, Zhang AY (2020) Prevalence and transmission of antimicrobial-resistant Staphylococci and Enterococci from shared bicycles in Chengdu. China Sci Total Environ 738:139735. https://doi.org/10.1016/j.scitotenv.2020.139735
Guo Y, Qiu T, Gao M, Sun Y, Cheng S, Gao H, Wang X (2021) Diversity and abundance of antibiotic resistance genes in rhizosphere soil and endophytes of leafy vegetables: Focusing on the effect of the vegetable species. J Hazard Mater 415:12595. https://doi.org/10.1016/j.jhazmat.2021.125595
He LY, Ying GG, Liu YS, Su HC, Chen J, Liu SS, Zhao JL (2016) Discharge of swine wastes risks water quality and food safety: antibiotics and antibiotic resistance genes, from swine sources to the receiving environments. Environ Int 92–93:210–219. https://doi.org/10.1016/j.envint.2016.03.023
He LY, He LK, Liu YS, Zhang M, Zhao JL, Zhang QQ, Ying GG (2019) Microbial diversity and antibiotic resistome in swine farm environments. Sci Total Environ 685:197–207. https://doi.org/10.1016/j.scitotenv.2019.05.369
Hooban B, Fitzhenry K, Cahill N, Joyce A, O'Connor L, Bray JE, Brisse S, Passet V, Abbas Syed R, Cormican M, Morris D (2021) A point prevalence survey of antibiotic resistance in the Irish environment, 2018–2019. Environ Int 152:106466. https://doi.org/10.1016/j.envint.2021.106466
Hu T, Dai Q, Chen H, Zhang Z, Dai Q, Gu X, Yang X, Yang Z, Zhu L (2020) Geographic pattern of antibiotic resistance genes in the metagenomes of the giant panda. Microb Biotechnol. https://doi.org/10.1111/1751-7915.13655
Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. Bmc Bioinformatics 11(119). https://doi.org/10.1186/1471-2105-11-119
Jia S, Bian K, Shi P, Ye L, Liu CH (2020) Metagenomic profiling of antibiotic resistance genes and their associations with bacterial community during multiple disinfection regimes in a full-scale drinking water treatment plant. Water Res 176:115721. https://doi.org/10.1016/j.watres.2020.115721
Jia H, Chen Y, Wang J, Xie X, Ruan Z (2019) Emerging challenges of whole-genome-sequencing-powered epidemiological surveillance of globally distributed clonal groups of bacterial infections, giving Acinetobacter baumannii ST195 as an example. Int J Med Microbiol 309(7). https://doi.org/10.1016/j.ijmm.2019.151339
Jo H, Raza S, Farooq A, Kim J, Unno T (2021) Fish farm effluents as a source of antibiotic resistance gene dissemination on Jeju Island. South Korea Environ Pollut 276:116764. https://doi.org/10.1016/j.envpol.2021.116764
Joyce A, McCarthy CGP, Murphy S, Walsh F (2019) Antibiotic resistomes of healthy pig faecal metagenomes. Microb Genom 5(5). https://doi.org/10.1099/mgen.0.000272
Kolde R (2019) Implementation of heatmaps that offers more control over dimensions and appearance. https://CRAN.R-project.org/package=pheatmap
Li B, Ju F, Cai L, Zhang T (2015) Profile and fate of bacterial pathogens in sewage treatment plants revealed by high-throughput metagenomic approach. Environ Sci Technol 49(17):10492–10502. https://doi.org/10.1021/acs.est.5b02345
Li D, Liu C-M, Luo R, Sadakane K, Lam T-W (2015) MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31(10):1674–1676. https://doi.org/10.1093/bioinformatics/btv033
Li J, Bi Z, Ma S, Chen B, Cai C, He J, Schwarz S, Sun C, Zhou Y, Yin J, Hulth A, Wang Y, Shen Z, Wang S, Wu C, Nilsson LE, Walsh TR, Borjesson S, Shen J, Sun Q, Wang Y (2019) Inter-host transmission of carbapenemase-producing Escherichia coli among humans and backyard animals. Environ Health Perspect 127(10):107009. https://doi.org/10.1289/EHP5251
Li H, Kang Z, Jiang E, Song R, Zhang Y, Qu G, Wang T, Jia H, Zhu L (2021) Plasma induced efficient removal of antibiotic-resistant Escherichia coli and antibiotic resistance genes, and inhibition of gene transfer by conjugation. J Hazard Mater 419:126465. https://doi.org/10.1016/j.jhazmat.2021.126465
Liu L, Wang H, Xu W, Yin Y, Ren Y, Bu H, Zhao D (2022) Tracking Clostridium perfringens strains from breeding duck farm to commercial meat duck farm by multilocus sequence typing. Vet Microbiol 266:109356. https://doi.org/10.1016/j.vetmic.2022.109356
Mahaney AP, Franklin RB (2022) Persistence of wastewater-associated antibiotic resistant bacteria in river microcosms. Sci Total Environ 819:153099. https://doi.org/10.1016/j.scitotenv.2022.153099
Mao Y, Akdeniz N, Nguyen TH (2021) Quantification of pathogens and antibiotic resistance genes in backyard and commercial composts. Sci Total Environ 749:149197. https://doi.org/10.1016/j.scitotenv.2021.149197
Mencia-Ares O, Cabrera-Rubio R, Cobo-Diaz JF, Alvarez-Ordonez A, Gomez-Garcia M, Puente H, Cotter PD, Crispie F, Carvajal A, Rubio P, Arguello H (2020) Antimicrobial use and production system shape the fecal, environmental, and slurry resistomes of pig farms. Microbiome 8(1):164. https://doi.org/10.1186/s40168-020-00941-7
Munk P, Knudsen BE, Lukjancenko O, Duarte ASR, Van Gompel L, Luiken REC, Smit LAM, Schmitt H, Garcia AD, Hansen RB, Petersen TN, Bossers A, Ruppé E, Graveland H, van Essen A, Gonzalez-Zorn B, Moyano G, Sanders P, Chauvin C, David J, Battisti A, Caprioli A, Dewulf J, Blaha T, Wadepohl K, Brandt M, Wasyl D, Skarzyńska M, Zajac M, Daskalov H, Saatkamp HW, Stärk KDC, Lund O, Hald T, Pamp SJ, Vigre H, Heederik D, Wagenaar JA, Mevius D, Aarestrup FM, Group E (2018) Abundance and diversity of the faecal resistome in slaughter pigs and broilers in nine European countries. Nat Microbiol 3(8):898–908. https://doi.org/10.1038/s41564-018-0192-9
Neuwirth E (2014) RColorBrewer: ColorBrewer Palettes. R package version. https://CRAN.R-project.org/package=RColorBrewer
Oksanen J BF, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2018) vegan: community ecology package. R package version. https://CRAN.R-project.org/package=vegan
O'Neill J (2014) Antimicrobial resistance: tackling a crisis for the health and wealth of nations. https://wellcomecollection.org/works/rdpck35v
Phuong Hoa PT, Nonaka L, Hung Viet P, Suzuki S (2008) Detection of the sul1, sul2, and sul3 genes in sulfonamide-resistant bacteria from wastewater and shrimp ponds of north Vietnam. Sci Total Environ 405(1–3):377–384. https://doi.org/10.1016/j.scitotenv.2008.06.023
Pınar Ş (2021) Prevalence, antibiotic resistance, and enterotoxin production of Staphylococcus aureus isolated from retail raw beef, sheep, and lamb meat in Turkey. Int J Food Microbiol. https://doi.org/10.1016/j.ijfoodmicro.2021.109461
Rodríguez EA, Ramirez D, Balcázar JL, Jiménez JN (2021) Metagenomic analysis of urban wastewater resistome and mobilome: a support for antimicrobial resistance surveillance in an endemic country. Environ Pollut 276. https://doi.org/10.1016/j.envpol.2021.116736
Song L, Wang C, Jiang G, Ma J, Li Y, Chen H, Guo J (2021) Bioaerosol is an important transmission route of antibiotic resistance genes in pig farms. Environ Int 154:106559. https://doi.org/10.1016/j.envint.2021.106559
Tang X, Lou C, Wang S, Lu Y, Liu M, Hashmi MZ, Liang X, Li Z, Liao Y, Qin W, Fan F, Xu J, Brookes PC (2015) Effects of long-term manure applications on the occurrence of antibiotics and antibiotic resistance genes (ARGs) in paddy soils: evidence from four field experiments in south of China. Soil Biol Biochem 90:179–187. https://doi.org/10.1016/j.soilbio.2015.07.027
von Meijenfeldt FAB, Arkhipova K, Cambuy DD, Coutinho FH, Dutilh BE (2019) Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol 20(1):217. https://doi.org/10.1186/s13059-019-1817-x
Wang Y, Zhang R, Li J, Wu Z, Yin W, Schwarz S, Tyrrell JM, Zheng Y, Wang S, Shen Z, Liu Z, Liu J, Lei L, Li M, Zhang Q, Wu C, Zhang Q, Wu Y, Walsh TR, Shen J (2017) Comprehensive resistome analysis reveals the prevalence of NDM and MCR-1 in Chinese poultry production. Nat Microbiol 2:16260. https://doi.org/10.1038/nmicrobiol.2016.260
Wang J, Gu J, Wang X, Song Z, Dai X, Guo H, Yu J, Zhao W, Lei L (2021) Enhanced removal of antibiotic resistance genes and mobile genetic elements during swine manure composting inoculated with mature compost. J Hazard Mater 411:125135. https://doi.org/10.1016/j.jhazmat.2021.125135
Wang M-G, Zhang R-M, Wang L-L, Sun R-Y, Bai S-C, Han L, Fang L-X, Sun J, Liu Y-H, Liao X-P (2021) Molecular epidemiology of carbapenemase-producing Escherichia coli from duck farms in south-east coastal China. J Antimicrob Chemother 76(2):322–329. https://doi.org/10.1093/jac/dkaa433
Wang X, Lin D, Huang Z, Zhang J, Xie W, Liu P, Jing H, Wang J (2021) Clonality, virulence genes, and antibiotic resistance of Staphylococcus aureus isolated from blood in Shandong. China BMC Microbiol 21(1):281. https://doi.org/10.1186/s12866-021-02344-6
Wang G, Kong Y, Yang Y, Ma R, Li L, Li G, Yuan J (2022) Composting temperature directly affects the removal of antibiotic resistance genes and mobile genetic elements in livestock manure. Environ Pollut 303:119174. https://doi.org/10.1016/j.envpol.2022.119174
Wang C, Li P, Yan Q, Chen L, Li T, Zhang W, Li H, Chen C, Han X, Zhang S, Xu M, Li B, Zhang X, Ni H, Ma Y, Dong B, Li S, Liu S (2019) Characterization of the pig gut microbiome and antibiotic resistome in industrialized feedlots in China. mSystems 4(6). https://doi.org/10.1128/mSystems.00206-19
Wang M-G, Yu Y, Wang D, Yang R-S, Jia L, Cai D-T, Zheng S-L, Fang L-X, Sun J, Liu Y-H, Liao X-P (2021) The emergence and molecular characteristics of New Delhi metallo β-lactamase-producing Escherichia coli from ducks in Guangdong, China. Front Microbiol 12(1678). https://doi.org/10.3389/fmicb.2021.677633
Wichmann F, Udikovic-Kolic N, Andrew S, Handelsman J (2014) Diverse antibiotic resistance genes in dairy cow manure. mBio 5(2):e01017. https://doi.org/10.1128/mBio.01017-13
Wood DE, Lu J, Langmead B (2019) Improved metagenomic analysis with Kraken 2. Genome Biology 20(1):257. https://doi.org/10.1186/s13059-019-1891-0
Xiong W, Wang Y, Sun Y, Ma L, Zeng Q, Jiang X, Li A, Zeng Z, Zhang T (2018) Antibiotic-mediated changes in the fecal microbiome of broiler chickens define the incidence of antibiotic resistance genes. Microbiome 6(1):34. https://doi.org/10.1186/s40168-018-0419-2
Xiu L, Liu Y, Wu W, Chen S, Zhong Z, Wang H (2020) Prevalence and multilocus sequence typing of Clostridium perfringens isolated from 4 duck farms in Shandong province. China Poult Sci 99(10):5105–5117. https://doi.org/10.1016/j.psj.2020.06.046
Xu X, Ma W, Zhou K, An B, Huo M, Lin X, Wang L, Wang H, Liu Z, Cheng G, Huang L (2022) Effects of composting on the fate of doxycycline, microbial community, and antibiotic resistance genes in swine manure and broiler manure. Sci Total Environ 832:155039. https://doi.org/10.1016/j.scitotenv.2022.155039
Yang T, Jiang L, Han Y, Liu J, Wang X, Yan X, Liu J (2020) Linking aerosol characteristics of size distributions, core potential pathogens and toxic metal(loid)s to wastewater treatment process. Environ Pollut 264:114741. https://doi.org/10.1016/j.envpol.2020.114741
Yin X, Jiang X-T, Chai B, Li L, Yang Y, Cole JR, Tiedje JM, Zhang T (2018) ARGs-OAP v2.0 with an expanded SARG database and Hidden Markov models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics 34(13):2263–2270. https://doi.org/10.1093/bioinformatics/bty053
Zeng J, Pan Y, Yang J, Hou M, Zeng Z, Xiong W (2019) Metagenomic insights into the distribution of antibiotic resistome between the gut-associated environments and the pristine environments. Environ Int 126:346–354. https://doi.org/10.1016/j.envint.2019.02.052
Zhang H, Chang F, Shi P, Ye L, Zhou Q, Pan Y, Li A (2019) Antibiotic resistome alteration by different disinfection strategies in a full-scale drinking water treatment plant deciphered by metagenomic assembly. Environ Sci Technol 53(4):2141–2150. https://doi.org/10.1021/acs.est.8b05907
Zhang H, Chen S, Zhang Q, Long Z, Yu Y, Fang H (2020) Fungicides enhanced the abundance of antibiotic resistance genes in greenhouse soil. Environ Pollut 259:113877. https://doi.org/10.1016/j.envpol.2019.113877
Zhang H, Zhang Z, Song J, Cai L, Yu Y, Fang H (2020) Foam shares antibiotic resistomes and bacterial pathogens with activated sludge in wastewater treatment plants. J Hazard Mater 408:124855. https://doi.org/10.1016/j.jhazmat.2020.124855
Zhang Y, Zheng Y, Zhu Z, Chen Y, Dong H (2020) Dispersion of antibiotic resistance genes (ARGs) from stored swine manure biogas digestate to the atmosphere. Sci Total Environ. https://doi.org/10.1016/j.scitotenv.2020.144108
Zhang R-M, Liu X, Wang S-L, Fang L-X, Sun J, Liu Y-H, Liao X-P (2021) Distribution patterns of antibiotic resistance genes and their bacterial hosts in pig farm wastewater treatment systems and soil fertilized with pig manure. Sci Total Environ 758:143654–143654. https://doi.org/10.1016/j.scitotenv.2020.143654
Zhao R, Yu K, Zhang J, Zhang G, Huang J, Ma L, Deng C, Li X, Li B (2020) Deciphering the mobility and bacterial hosts of antibiotic resistance genes under antibiotic selection pressure by metagenomic assembly and binning approaches. Water Res 186:116318. https://doi.org/10.1016/j.watres.2020.116318
Funding
This work was jointly supported by the National Natural Science Foundation of China (31730097), the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (32121004), and Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2019BT02N054).
Author information
Authors and Affiliations
Contributions
KL and MW analyzed the data and wrote the manuscript. YZ and CF visualized the data. RZ, LF, and JS revised and reviewed the manuscript. YL reviewed the manuscript and supervised the project. XL designed and supervised the project. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, K., Wang, M., Zhang, Y. et al. Distribution of antibiotic resistance genes and their pathogen hosts in duck farm environments in south-east coastal China. Appl Microbiol Biotechnol 108, 136 (2024). https://doi.org/10.1007/s00253-023-12842-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00253-023-12842-4