
Abstract
Serology assays are essential tools to mitigate the effect of COVID-19, help to identify previous SARS-CoV-2 infections or vaccination, and provide data for surveillance and epidemiologic studies. In this study, we report the production and purification process of the receptor-binding domain (RBD) of SARS-CoV-2 in HEK293 cells, which allowed the design, optimization, and validation of an indirect ELISA (iELISA) for the detection of human anti-RBD antibodies. To find the optimal conditions of this iELISA, a multivariate strategy was performed throughout design of experiments (DoE) and response surface methodology (RSM), one of the main tools of quality by design (QbD) approach. The adoption of this strategy helped to reduce the time and cost during the method development stage and to define an optimum condition within the analyzed design region. The assay was then validated, exhibiting a sensitivity of 94.24 (86.01–98.42%; 95% CI) and a specificity of 95.96% (89.98–98.89%; 95% CI). Besides, the degree of agreement between quality results assessed using kappa’s value was 0.92. Hence, this iELISA represents a high-throughput technique, simple to perform, reliable, and feasible to be scaled up to satisfy the current demands. Since RBD is proposed as the coating antigen, the intended use of this iELISA is not only the detection of previous exposure to the virus, but also the possibility of detecting protective immunity.
Key points
• RBD was produced in 1-L bioreactor and highly purified.
• An iELISA assay was optimized applying QbD concepts.
• The validation procedure demonstrated that this iELISA is accurate and precise.
Graphical Abstract

Similar content being viewed by others

Introduction
During the first 2 years of pandemic, COVID-19 disease, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has altered normal living conditions with significant health and social impact, debilitating the economy globally. COVID-19 has provoked more than 6.5 million deaths by October 10, 2022 (WHO 2022), with symptoms ranging from fever, dry cough, tiredness, loss of taste, smell and speech, sore throat, and chest pain, to breathing difficulties (Agrahari et al. 2021).
The SARS-CoV-2 genome is a non-segmented large positive-sense stranded RNA, encoding 29 proteins: 25 putative non-structural and accessory proteins and four structural proteins (Wu et al. 2022). The structural proteins, essential for virus assembly, include spike (S), membrane (M), 16S nucleo-capsid (N), and envelope (E) proteins (Agrahari et al. 2021). The S protein has two subunits, the S1 and S2, respectively. The S1 subunit is mainly responsible for receptor binding, as it contains the receptor-binding domain (RBD) at its C-terminus. The RBD domain recognizes the host receptor angiotensin-converting enzyme 2 (ACE2) to facilitate viral entry into epithelial cells. The S2 subunit forms the stalk that provides spike structure, since it contains multi-components, such as the hydrophobic fusion peptide, heptad repeats, and transmembrane domain (Agrahari et al. 2021; Wu et al. 2022).
Testing strategies are essential tools to mitigate the effect of COVID-19, help to diagnose the disease, and provide data for surveillance and epidemiologic studies. Also, they are critical during development and monitoring stages of different vaccine designs and they help to identify possible convalescent plasma donors (Bonanni et al. 2021; Liu and Rusling 2021). Currently, nucleic acid amplification tests (NAATs), and most commonly the RT-PCR, are the gold standard for the diagnosis of the disease. Virus neutralization tests (VNTs) are the gold standard of serology assays, detecting the functional activities of antibodies, that is, their neutralization capacity to block the entrance of the virus into the host cells (Fernández-Barat et al. 2020; Bonanni et al. 2021). However, VNTs exhibit several drawbacks, including the requirement of containment facilities and the difficulty of standardization. Hence, alternative serology methods have been developed, such as immunofluorescent assay (IFA), enzyme-linked immunosorbent assay (ELISA), and western blot. Among them, ELISA seems to be the more versatile assay, demonstrating good performance in terms of sensitivity and specificity (Alandijany et al. 2020; Faizo et al. 2021; Krähling et al. 2021; Ayón-Núñez et al. 2022; Fernandes-Siqueira et al. 2022).
In this study, we report the production and purification process of RBD in HEK293 cells, which allowed the design, validation, and optimization of an indirect ELISA (iELISA) for the detection of human anti-RBD antibodies through the use of QbD tools.
Materials and methods
RBD sequence design and plasmid construction
The RBD sequence was obtained from the NCBI Reference Sequence for SARS-CoV-2 (residues 329 to 522). It was fused to the native signal peptide of the spike protein (residues 1 to 14) and to a 6 × His-tag at the C-terminal region (Fig. 1A). Sequences were designed according to the information reported in the bibliography for the first virus isolate, Wuhan-Hu-1 (GenBank accession number: OP160527.1).

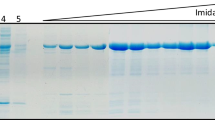
RBD production and purification. A Scheme of RBD protein. Natural signal peptide (1–14), RBD domain (329–522), and C-terminal His-tag. B Transient expression of RBD in sHEK293 cells. Supernatants were collected 48 h after transfection and analyzed by SDS-PAGE/western blot using an anti-SARS-Cov-2 spike neutralizing mAb for detection. C SDS-PAGE following Coomassie brilliant blue of purified RBD profile. Lane 1: molecular mass protein standards (Precision Plus Protein, Bio-Rad). D SDS-PAGE silver staining of purified RBD profile. Lane 1: molecular mass protein standards (Precision Plus Protein, Bio-Rad)
Briefly, the RBD DNA sequence was codon-optimized for mammalian cell expression and obtained by chemical synthesis (Gene Universal Inc., Newark DE, USA). The DNA sequence was cloned under the CAG promoter sequence, which is composed by a CMV enhancer followed by the chicken β-actin promoter and a chimeric intron (Niwa et al. 1991). The backbone, pLV expression plasmid, was previously described (Mufarrege et al. 2014). It contains a puromycin resistance gene for recombinant cell line generation in animal cells. The expression plasmid was amplified and purified from competent E. coli cells. Its sequence was verified by dideoxy DNA sequencing (Macrogen, Seoul, South Korea).
RBD transfection and cell line generation
Before starting the transfection protocol, suspension adapted-HEK293 cells (sHEK) were cultured in transfection medium (DMEM [Gibco] plus glutamine 2 mM) under adherent growth conditions (10% fetal bovine serum [FBS]) during 48 h. Two rounds of 3 transfections each were performed, rendering Tf6 cell line, as described below. Initially, 2 × 105 cell.mL−1 were cultured in 6-well plates at 37 °C and 5% CO2. After 24 h, plasmid/lipid (Lipofectamine™ 3000 Transfection Reagent [Invitrogen]) complexes were formulated and added to the cells, following the transfection procedure suggested by the manufacturer. Particularly, a 1:2 plasmid/lipid ratio was used. At 48 h post-transfection, cell supernatants were harvested and analyzed by SDS-PAGE/western blot to verify RBD expression. Then, transfected cells were seeded again as previously described (2 × 105 cell.mL−1), to proceed with the same transfection protocol. The first round was completed after three consecutive rounds of transfections (Tf3 cell line). Selective pressure with 5 µg.mL−1 puromycin was performed in order to allow only the growth of the recombinant cells, i.e., the ones that stably express the puromycin resistance and RBD. Thereafter, the second round of transfections was performed. In this way, Tf3 cell line treated with 5 µg.mL−1 puromycin was transfected 3 more times (Tf6 cell line), following the same protocol described above. Then, the Tf6 cell culture media were supplemented with different concentrations of puromycin (5, 10, and 50 µg.mL−1) to allow only the growth of the recombinant cells. Then, Tf6 cell line was cultured without antibiotic selection to evaluate the productivity, and supernatants were analyzed by SDS-PAGE/western blot.
The sHEK cell line that exhibited the highest productivity of RBD was gradually adapted to a chemically defined serum-free medium (CD BHK-21 Production Medium, Thermo Fisher). Specifically, 50% of the adherent growth medium was initially replaced by serum-free medium. When cell growth was observed, a 75:25% serum-free medium/adherent growth medium was used for cell culture. Subsequently, 85:15% and 95:5% ratios were employed, and finally, 100% serum-free medium was used. Afterwards, RBD productivity was analyzed by SDS-PAGE/western blot.
RBD production
Suspension RBD cell line was grown in Erlenmeyer flasks (Corning, USA) agitated at 140 rpm in a shaker incubator (CERTOMAT®CTplus, Sartorius, Germany). Afterwards, cells were transferred to a 1-L bioreactor operated in perfusion mode (BIOSTAT® Qplus, Sartorius), and cultured using the CD BHK-21 Production Medium. The culture parameters were controlled on-line, maintaining a pH of 7.2, a pO2 of 30% air saturation, and an agitation rate of 140 rpm. Temperature was varied between 37 and 33 °C. Sample collection was performed daily to determine cell density, cell viability, and glucose and lactate levels. Also, RBD was monitored by SDS-PAGE/western blot.
RBD purification
RBD protein was purified from clarified culture supernatants by immobilized metal affinity chromatography (IMAC) using a highly cross-linked agarose with a covalently coupled chelating group (IMAC Sepharose 6 Fast flow, Cytiva), previously activated with NiCl2. Clarified supernatants were obtained by filtration with 0.22-µm membrane filters (Millipore Express, Merck). Conditioned sample was loaded onto an IMAC resin packed in a XK 16/20 column connected to an AKTA explorer 100 Air (GE Healthcare), previously equilibrated with 20 mM imidazole, 0.5 M NaCl, 20 mM sodium phosphate buffer, and pH 7.4. A first wash step was performed with the same equilibration buffer, followed by a second wash step carried out with 50 mM imidazole, 0.5 M NaCl, 20 mM sodium phosphate buffer, and pH 7.4. The RBD protein was eluted using an isocratic 250 mM imidazole gradient in the same buffer. Fractions containing the protein were pooled, dialyzed, and concentrated against storage buffer (PBS, phosphate buffer saline). Finally, purified RBD was aliquoted and stored at − 70 °C until used.
RBD characterization
Protein measurement; absorbance at 280 nm
Concentration of purified RBD samples was estimated spectrophotometrically using absorbance at 280 nm (33,462 M−1 cm−1). A spectrophotometer Ultrospect 1000 Pro UV/Vis (Amersham Bioscience) with a quartz cuvette of a 1-cm path length was employed. The protein buffer formulation (PBS) was employed as a blank control. The extension coefficient was predicted directly from RBD sequence.
SDS-PAGE and western blot analysis
Protein sample electrophoresis profiles were analyzed using a 12% (w/v) polyacrylamide resolving gel and 5% (w/v) stacking gel. Separation was performed at 200 V for 65 min. Samples were mixed with 4 × sample buffer (2% [w/v] SDS, 10% glycerol, 50 mM Tris–HCl pH 6.8, 0.05% [v/v] bromophenol blue), in reducing (5% [v/v] beta-mercaptoethanol) and non-reducing conditions.
Gels were stained with Coomassie blue and destained with a solution containing 15% (v/v) methanol and 10% (v/v) acetic acid.
Also, silver staining was performed, according to the following sequential phases: 1, protein fixation (50% v/v methanol, 12% [v/v] acetic acid, and 0.05% [v/v] formaldehyde, during 1 h at room temperature); 2, sensitization (0.2 g.L−1 sodium thiosulfate solution, during 1 min); 3, silver impregnation (2 g.L−1 silver nitrate, 0.075% [v/v] formaldehyde solution, during 20 min); and 4, image development (60 g.L−1 sodium carbonate, 0.05% [v/v] formaldehyde, and 4 mg.L−1 sodium thiosulfate solution).
For western blot analysis, proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad Laboratories Inc.). Membranes were blocked with Tris-buffered saline (TBS, 50 mM Tris–HCl, pH 7.5, 150 mM NaCl), 5% (w/v) non-fat milk. Recombinant proteins were detected with the pair anti-SARS-Cov-2 spike neutralizing monoclonal antibody (mAb) (Creative Diagnostics) at 1:1000 and HRP-conjugated rabbit anti-mouse (ISAL, FBCB, UNL) at 1: 750. The reaction was visualized using a chemiluminescence reagent (Pierce® ECL Western Blotting Substrate, Thermo Scientific).
The relative quantification of bands was performed using the free-image processing Image J software (http://fiji.sc/#download).
Analytical reverse phase (RP-) high-performance liquid chromatography (HPLC)
Ten micrograms of purified RBD samples was analyzed by RP-HPLC method using a Jupiter C4 Phenomenex column (250 mm × 4.6 mm, 300 Å-pore size, and 5-µm particle diameter). Solutions A (Milli-Q water 0.1% trifluoroacetic (TFA)) and B (acetonitrile, 0.1% TFA, HPLC grade, Merk) were employed in a 35-min gradient, increasing the percentage of B from 20 to 80, and a 10-min isocratic mode with 100% of B phase. The injected sample volume was 100 µL and the column temperature was maintained at 25 °C. Elution profiles were recorded at 210 nm under a constant flow rate of 1 mL/min.
Intrinsic fluorescent spectroscopy
Spectrofluorimetric measurements were performed using a PerkinElmer LS-55 luminescence spectrometer equipped with a Xenon discharge lamp, Monk-Gillieson type monochromators, and a gated photomultiplier. The acquisition of data was performed using the FL Winlab v4.00.03 software supplied by PerkinElmer.
In all cases, quartz cells with planar walls of 100 µL were used. A 5-nm slit width was used in excitation and 7 nm in emission monochromators; a 650 V detector voltage and a scan speed of 600 nm.min−1 were set. Emission spectra were recorded from 290 to 500 nm in 1-nm steps with excitation at 275 nm. The Trp emission spectra, employed as a positive control, were recorded from 310 to 500 nm in 1-nm steps with excitation at 295 nm.
Urea and DTT treatment
Purified RBD was treated with 6 M urea for 1 h at 37 °C. Besides, its reduced form was prepared by incubating the samples with 100 mM DTT for 1 h at 37 °C. Aliquots of freshly stocked solution of urea and DTT were always used for the treatment.
PNGase F treatment
Five micrograms of purified RBD was mixed with 15 IU of PNGase F (New England Biolabs, UK) in a total volume of 20 µL in denaturing conditions and incubated overnight at 37 °C. Samples were analyzed by SDS-PAGE followed by Coomassie blue staining to verify the removal of glycans of the samples, as described above.
RBD-specific binding with HEK293T cells expressing hACE2
The specific interaction between RBD and ACE2 receptor was determined using HEK-ACE2 cells. HEK293T cells were used as a negative interaction control. Initially, a 24-well plate was incubated with 10 µg.mL−1 polyethyleneimine (PEI) in 150 mM NaCl during 2 h at 37 °C. This treatment favors the attachment of the cells to the plate and avoids their detachment during the procedure. Each well was washed with PBS to remove PEI. Then, 2 × 105 cells (HEK293T and HEK-ACE2) were seeded in DMEM 10% FBS. After 24 h, HEK293T and HEK-ACE2 monolayers were washed three times with PBS and incubated 45 min with 100 µg of RBD in PBS. Then, cells were washed again as previously described and incubated with 200 µL of a 1:1000 dilution of an in-house polyclonal rabbit anti-RBD (previously produced using standard protocols (Harlow and Lane 1988)). After a 30-min incubation step at room temperature, cells were washed again and 200 µL of Alexa Fluor 488-conjugated goat anti-mouse antibody (Thermo Fisher Scientific) diluted 1:500 was added to the cells. Incubation was performed at room temperature in the dark for 30 min. To stain the nucleus of the cells, 200 µL of PBS containing Hoechst 1 µg.mL−1 (Thermo Fisher Scientific) was used. Finally, cells were washed, and analyzed by fluorescence microscopy (Eclipse Ti–S, Nikon Instruments Inc.).
Samples
For this study, 99 negative prepandemic samples and 70 positive samples were evaluated. Fifty prepandemic serum samples collected between October 2018 and December 2019 were provided by CUDAIO (Centro Único de Donación, Ablación e implante de Órganos, Santa Fe, Argentina). In addition, 49 prepandemic samples were collected at Laboratorio Central de la Provincia de Santa Fe (Santa Fe, Argentina) before 2019. Regarding positive samples, 37 were obtained at CEMAFE Hospital (Santa Fe, Argentina) and confirmed by the anti-spike chemiluminescence assay or RT-PCR. Besides, and 33 COVID-19 positive plasma samples were collected at Alkemy Diagnostico (Santa Fe, Argentina) and confirmed by Elecsys® anti-SARS-CoV-2 RBD (Diagnostics Roche). One of the positive samples (identified as 1# ( +)) and two prepandemic sera (named 2# and 3# ( −)) were used as positive and negative controls, respectively, for the design and optimization of the iELISA.
In addition, 46 sera from healthy patients were analyzed to evaluate seroconversion after the first and second doses of a complete vaccination schedule. Human serum samples were stored at − 20 °C until required.
Assays involving human samples were approved and supervised by the Advisory Committee on Ethics and Security of the School of Biochemistry and Biological Sciences, Universidad Nacional del Litoral, according to international guidelines (Council for International Organizations of Medical Sciences (CIOMS), ISBN 92–9036-075–5).
Optimized iELISA
Ninety-six-well plates were coated with 100 µL per well of 136.3 ng of purified RBD in 50 mM carbonate/bicarbonate buffer (pH 9.6) and incubated overnight (ON) at 4 °C. After blocking 1 h at 37 °C with 5% (w/v) non-fat milk in PBS, plates were incubated with 1:40 diluted serum or plasma sample for 1 h at 37 °C. At the same time, the following controls were tested in each run: (1) a blank reactive control (BRC); (2) a negative sample control (prepandemic serum); and (3) an antigen-free control (without RBD) and a 1:40 positive control during the sample incubation step (named control A). Then, plates were incubated with a 1:3225 diluted rabbit anti-human IgG-HRP (Santa Cruz Biotechnology) for 1 h at 37 °C. Finally, plates were incubated for 13 min with substrate solution (0.5 mg/mL o-phenylenediamine, 0.12% (v/v) H2O2 in 50 mM phosphate citrate buffer). Absorbance was measured at 492 nm with a microtiter plate reader (LabSystems Multiskan, Thermo Fisher Scientific). Between every step, plates were washed six times with PBS, 0.05% (v/v) Tween-20 (PBS-T). Dilutions of tested samples and antibodies were prepared in PBS-T containing 0.2% (w/v) non-fat milk.
The same procedure was applied for the iELISA employing stressed RBD sample treated with 100 mM DTT as coating antigen.
iELISA optimization
To find the optimal iELISA assay conditions that allow obtaining the highest ratio between the signals of the positive and negative sera, and also the maximum difference with the control conditions, a DoE and RSM approach were applied. The multivariate DoE and RSM strategies consume less time and resources than the traditional univariate procedure (one-variable-at-a-time, OVAT) for the same purpose (Goicoechea 2018). In these approaches, the effects of several factors are studied simultaneously. Varying the levels of all factors involved in the process instead of one at a time allows the study of the interactions between the factors. Here, a three-level full factorial design was performed, with two factors (amount of coating antigen and dilution of rabbit anti-human IgG-HRP) and four center points. Each factor level was selected based on preliminary experiments results. Thirteen runs were carried out. The evaluated responses (signal ratio positive/negative sera (P/N), signal ratio negative sera/control A; signal ratio negative sera/BRC; signal ratio positive sera/control A; and signal ratio positive sera/BRC) were fitted to hierarchical polynomial models, once the outliers were eliminated (Table 1). Then, the model coefficients were estimated by backward multiple regression, and validated by the ANOVA method (analysis of variance, p < 0.05). During the response’s optimization stage, the desirability function was applied (Derringer y Suich 1980). The experiments were performed in a randomized order to ensure the independence of the results. Finally, the predicted optimum condition was validated. Stat-Ease Design-Expert Version 7.0.3 was applied to analyze the design’s results.
iELISA validation
A preliminary cut-off value was estimated as the mean of the signal of negative sera (prepandemic sera, n = 81) + three standard deviations (\(\stackrel{\mathrm{-}}{\text{x}}\) ± 3 × SD). Likewise, the estimated sensitivity and specificity were determined, evaluating 81 and 26 negative and positive serum samples, respectively.
Then, receiver-operating characteristic (ROC) analysis was performed on the iELISA signal data to determine the optimal cut-off, based on Youden index, and the area under the curve (AUC) to assess the method accuracy. The assay’s performance was assessed estimating sensitivity, specificity, and positive and negative predictive values, using contingency table analysis procedure. The confidence intervals were calculated following Wilson/Brown method. GraphPad Prism 5 was employed to analyze the results. Agreement between positive samples, evaluated by the reference methods (Elecsys®, RT-PCR), and the developed iELISA was assessed using Cohen’s kappa statistic.
Precision was evaluated at two levels—repeatability and intermediate precision—and it was analyzed by inter- and intra-coefficient of variation (CV %). Three positive serum samples (high, medium, and low signal serum) were evaluated in triplicate in three independent assays.
Results
RBD expression and production
In this study, to obtain the recombinant version of RBD, expression plasmids were used for transient transfections of sHEK cells under adherent conditions. Thereafter, supernatants of transiently transfected sHEK cells were analyzed by SDS-PAGE followed by western blot to assess protein identity. A protein band at 30 kDa confirmed the expression of the RBD protein (Fig. 1B). sHEK Tf6 RBD cell line treated with 5 µg.mL−1 of puromycin exhibited the highest RBD productivity. Hence, this cell line was gradually adapted to the chemically defined serum-free medium. This process took approximately 15 days, until normal daily growth was observed.
Thereafter, cells were cultured in serum-free production medium in a 1-L bioreactor in perfusion mode during 10 days, reaching cell densities up to 1.4 × 107 cell.mL−1. Perfusion rate varied between 0.25 and 1.0 reactor volumes per day, as it was required, maintaining nearly constant lactate and glucose amounts. A total volume of 12.3 L was harvested.
RBD purification
Clarified crude supernatant was loaded onto an IMAC resin, as RBD included a C-terminal His-tag. The RBD protein was efficiently bound to the resin, since no leakage of the protein was observed during the loading and first washing step. A small percentage of RBD protein was lost during the second wash step; nonetheless, it helped to improve the purity profile. RBD eluted with high purity levels (> 95%), as judged by SDS-PAGE analysis following by Coomassie brilliant blue and silver staining (Fig. 1C and D, respectively).
RBD characterization
The purified RBD sample expressed in HEK293 cells was characterized by RP-HPLC, fluorescent spectroscopy, and PNGase F treatment followed by SDS-PAGE and microscope fluorescence (for evaluation of RBD-ACE2 interaction) (Fig. 2).

RBD characterization. RP-HPLC analysis of A RBD and B RBD reduced form. C Steady-state fluorescent profile (at 275 nm excitation) of 8.5 µM of RBD (black), RBD treated with 6 M urea (red), and RBD treated with 100 mM DTT (blue). The Trp emission profile used as positive control is shown in green. D SDS-PAGE analysis of RBD protein treated ( +) and untreated ( −) with PNGase F under denaturing conditions, with ( +) and without ( −) heat treatment, and in the absence ( −) and presence ( +) of a reducing agent (β-ME). Lane 7: PNGAse F band pattern (36 kDa); lane 8: molecular mass protein standards (Precision Plus Protein, Bio-Rad). E Fluorescence microscopy to study RBD binding to HEK293T cells expressing hACE2. HEK293T cells were used as a negative control. First, cells were incubated with RBD (100 µg), followed by an incubation with an anti-RBD antibody. Thereafter, a secondary antibody (Alexa Fluor 488-conjugated goat anti-mouse antibody) was used for detection. Nuclei were stained with Hoechst. Bar scale: 50 µm
The elution profile of the RBD RP-HPLC showed a single sharp peak (Fig. 2A) at 19 min, suggesting the presence of a single disulfide-bonded species. In addition, this result indicates the absence of structurally related impurities, such as protein dimers. In contrast, reduced RBD sample showed three peaks at 20.5, 20.8, and 33.8 min, respectively (Fig. 2B), which would indicate the importance of disulfide bridges in protein stabilization.
Besides, by fluorescent spectroscopy, untreated RBD sample exhibited an emission maximum at 341 nm, whereas RBD treated with 6 M urea exhibited a 9 nm red-shift (350 nm) and a 20% less emission intensity. The emission maximum of the reduced form was located at 344 nm and showed a 50% less emission intensity (Fig. 2C). Those changes in the Trp emission profile are consistent with protein unfolding (Volkin et al. 2002; Gourbatsi et al. 2018).
As it can be seen from the analysis by SDS-PAGE, the purified RBD protein migrated as a 30.4 kDa single band in non-reducing SDS-PAGE (Fig. 2D, lane 1), suggesting the existence of glycosylation as a post-translational modification, since its theoretical mass resulting from the amino acid sequence is 26 kDa.
After PNGase F treatment and analysis under reducing conditions (addition of β-ME) and heat treatment, a band pattern with lower molecular mass was observed, indicating the removal of N-glycans (Fig. 2D, lane 6). On the contrary, after PNGase F treatment, a characteristic aggregation pattern (with bands above 150 kDa) was observed under non-reducing conditions and no thermal treatment (Fig. 2D, lane 2), which would indicate the importance of glycans in the conformational stability of the protein. When the N-deglycosylated protein was analyzed under non-reducing conditions and heat treatment, a partial aggregation pattern was observed (Fig. 2D, lane 4). Hence, based on these results, not only hydrophobic interactions would be involved in the formation of aggregates, but also disulfide bonds.
Fluorescence microscopy showed that purified RBD interacted with the human ACE2 expressed in HEK293 cells but not with HEK293T (Fig. 2E).
iELISA design and optimization: use of RBD as capture antigen
The goal of the iELISA was to establish a simple and reliable assay to detect human anti-SARS-CoV-2 antibodies.
Both binding and functional (neutralizing) antibodies can be the target of detection. However, only functional antibodies block virus entry into the host cells, whereas binding antibodies indicate the presence of previous exposure to a pathogen or vaccination. It is reported that antibodies against the RBD protein may provide a higher sensitivity and specificity for diagnosis, as RBD is considered the main target to elicit neutralizing antibodies (Bonanni et al. 2021; Liu and Rusling 2021). Thus, in this assay, a highly purified RBD protein was employed as coating antigen, and a rabbit anti-human IgG-HRP as the detection system. Thus, the intended use of this iELISA is not only to indicate previous exposure to the virus (or vaccination) but it could also indicate acquired immunity.
An optimization procedure was developed to improve the ratio between the signal of the positive and the negative sera, and the difference with the control conditions. A three-level full factorial design consisting in 13 experiments was performed (Table 1). The factors were studied in the following ranges: 50 to 150 ng of RBD (A) and 1/4000 to 1/2000 of rabbit anti-human IgG-HRP (Table 1). As can be seen, most of the model terms were significant and the lack of fit was not significant for each response (p > 0.05). Each model exhibited low CV% and high adjusted R-squared values, indicating good relationship between experimental data and the fitted models (Table 2). Besides, in each obtained model, the assumptions of homoscedasticity, normality, and randomness of the residuals were fulfilled.
The criterion used to optimize the five responses simultaneously corresponded to maximizing each response, giving to the response P/N ratio more importance. Under this optimization criterion, the predicted optimum conditions were the following: 136.36 ng of RBD (A) and 1/3225 of rabbit anti-human IgG-HRP (B), with a desirability function of D = 0.76 (Fig. 3A). This predicted point was then validated through an independent assay, exhibiting a good model prediction (data not shown). A classical dose–response curve for the optimized iELISA conditions can be seen in Fig. 3B.

iELISA optimization. A Desirability function plot. Response surface plots of the desirability function when optimizing the two evaluated factors. B Dose–response curve of positive and negative samples under optimization conditions. BRC: blank reactive control. Control A: an antigen-free control (without RBD) and a 1:40 positive control during the sample incubation step. C Effect of RBD conformational state in the iELISA performance. Positive and negative samples were evaluated employing native RBD (no treatment) and stressed RBD (following treatment with 100 mM DTT). Serum samples were diluted 1:40. Error bars represent the SD of n: 3 measurements. Abs: absorbance (492 nm)
Furthermore, we demonstrated that the performance of the validated iELISA is influenced by RBD conformational state. The SARS-CoV-2 antibody reactivity was modified by the conformational state of the RBD since a signal decrease was observed when a stressed RBD sample (100 mM DTT) was employed as coating antigen (Fig. 3C).
iELISA validation
To assess the reliability of the developed iELISA, a validation procedure was performed. A preliminary cut-off was estimated as the mean plus three standard deviations of 81 negative prepandemic sera samples, also evaluating 26 positive sera samples (Fig. 4A). The estimated cut-off value was 0.33 Abs. Except for a single prepandemic serum, all the evaluated negative samples were tested negative. Under these conditions, the estimated sensitivity was 76.92% (56.35–91.10%; 95% confidence interval), and the specificity was 98.77% (93.31–99.97%; 95% confidence interval).

iELISA validation. A Preliminary cut-off value of the assay. Dashed lines represent the estimated cut-off value. B ROC curve of validate iELISA assay. C Distribution data of negative and positive samples evaluated during the validation procedure. Samples were processed as described in the “Materials and methods” section. Dotted lines show the cut-off determined by ROC analysis and Youden index. Seventy positive samples (light-blue dots) and 99 prepandemic samples (green squares) were evaluated. D SARS-CoV-2 anti-RBD antibodies of vaccinated donors detected using the validated iELISA. Three types of vaccine platforms were analyzed (first and second dose): inactivated virus (BBIBP-CorV Sinopharm, Beijing Institute of Biological Products), viral vector (ChAdOx1-s, Oxford/AstraZeneca; Sputnik V, National Center Gamaleya de Epidemiology and Microbiology), and a combination (viral vector + mRNA-1273, Moderna). No vaccination (empty dots) corresponds to a not vaccinated donor whose antibody levels were elicited by COVID natural infection. From the vaccinated donors, 25% recorded a history of positive COVID-19
Then, a ROC analysis was performed, evaluating 99 prepandemic sera samples and 70 COVID19 positive sera samples (Fig. 4B). To assess the assay accuracy, the AUC was calculated, obtaining a high value of 0.9882, which indicates a good performance of the iELISA. The cut-off value was set at 0.2560 Abs using the Youden index as criteria. Hence, samples were classified as positive and negative using the contingence table (Table 3 and Fig. 4C), and the iELISA parameters were estimated. The sensitivity of the assay was 94.24 (86.01–98.42%; 95% confidence interval [CI]), the specificity 95.96% (89.98–98.89%; 95% CI), the positive predictive value 94.29% (86.01–98.42%; 95% confidence interval), and the negative predictive value (89.98–98.89%; 95% confidence interval). Besides, the degree of agreement between quality results assessed using kappa’s value was 0.92, which suggests an almost perfect agreement between the 169 pair results.
To assess the repeatability and the intermediate precision of the iELISA, the intra- and inter-CV% values were estimated, obtaining a relatively good performance in terms of precision. Intra-assay CV (%) value was 4.6% (3.1–6.0%; 95% CI) and interassay CV (%) was 11.8% (2.7–20.9%; 95% CI) (Table S1).
iELISA application to test anti-SARS-CoV-2 (RBD) immunoglobulins in human plasma vaccinated donors
To evaluate the performance of the validated iELISA, 46 plasma samples from donors vaccinated with three types of vaccine platforms were analyzed: inactivated virus (BBIBP-CorV Sinopharm, Beijing Institute of Biological Products), viral vector (ChAdOx1-s, Oxford/AstraZeneca; Sputnik V, National Center Gamaleya de Epidemiology and Microbiology), and a combination (viral vector + mRNA-1273, Moderna). For this study, the vaccination schedule of all platforms was considered completed after the second dose. Percentage of donors with different vaccine designs was as follows: 43.75% with the inactivated virus vaccine, 46.875% with the inactivated viral DNA platform, 6.25% with a combination of two vaccines, and 3.125% with no vaccination record at the time of extraction but being a convalescent COVID patient. In addition, from the vaccinated donors, a 25% recorded a history of positive COVID-19. Samples were collected at least 15 days after the second vaccination dose. For donors that did not meet this criterion, a second extraction was performed at the appropriate time (Table 4). Regardless of the vaccine design, all patients may have developed an immune response upon completion of the vaccination schedule (two doses), since all the corresponding plasma samples exceeded the cut-off value (0.2560) (Fig. 4D).
Discussion
On 11 March 2020, the WHO declared the novel coronavirus outbreak a pandemic, and since then, a global effort to mitigate the impact of COVID-19 disease has been done. In this context, testing strategies can help to diagnose the infection and provide surveillance data to policy makers and healthcare professionals. Molecular, antigen, and serological tests are the three main tools currently available. In this way, RT-PCR represents the gold standard for diagnosis, and the serological tests, the more adequate tools to provide evidence of prior infection, to study seroconversion dynamics and to evaluate the possible acquired immunity (Fernández-Barat et al. 2020; Bonanni et al. 2021). Antigen detection tests exhibit shorter turnaround times and reduced cost, being adequate tools in field applications. However, they are less sensitive, and therefore, all negative results must be confirmed with the gold standard tool to guarantee the exact diagnosis. Among the serological tests, the neutralization assay is the gold standard for determining antibody efficacy (GeurtsvanKessel et al. 2020). However, it has several drawbacks, for instance, its high cost and the requirement of a level 3 biosafety laboratory. On the contrary, ELISAs are simpler and less expensive, and they can detect both binding and functional antibodies and they also provide consistent results (Premkumar et al. 2020; Bonanni et al. 2021; Liu and Rusling 2021). Besides, antibodies exhibit higher stability in comparison with viral RNA during sample collection, preparation, transport, and storage.
To date, serology assays are based on N and S proteins as well as their subunits. However, despite ELISAs based on the N protein as coating antigen are commercially available, recent evidence reveals that the detection of binding antibodies against N protein may not correlate with the presence of neutralizing antibodies (Bonanni et al. 2021). On the contrary, as RBD domain binds to the ACE2 receptor on epithelial cells, resulting in virus entry in the host cell (Bonanni et al. 2021; Ibrahim et al. 2021), it is considered to be possibly the main target to elicit a strong neutralizing immune response.
In this work, we expressed, produced, and highly purified the RBD domain in suspension HEK293 cells. Even though CHO-K1 cells constitute the workhorses for production in the biopharmaceutical industry, HEK293 cells have also been used in manufacturing processes of already approved biodrugs and they can support the expression of difficult-to-express (DTE) proteins where CHO cells are suboptimal. Since viral proteins could be considered DTE, HEK293 cells constitute the chosen host for cell line generation (Zhu 2012; Rodrigues et al. 2015; Schwarz et al. 2020; Pulix et al. 2021; Malm et al. 2022). In those cases, transient production constitutes a good alternative. However, stable cell lines are preferred for large-scale production since they provide recombinant proteins with a consistent quality. Also, the bioprocess can be easily scalable without affecting the productivity and the protein quality (O’Flaherty et al. 2020). The majority of the reports published to date describing the expression of RBD domain in HEK293 cells choose the transient production instead of the stable cell line generation (Stadlbauer et al. 2020; Castro et al. 2021; Mehdi et al. 2021; Mehalko et al. 2021; Coria et al. 2022; Gstöttner et al. 2022; Amanat et al. 2020).
Here, a recombinant stable sHEK293 cell line producing RBD was obtained after six transfection events and selection with puromycin. The sHEK Tf6 RBD cell line was cultured in a 1-L bioreactor in perfusion mode, reaching high cell densities which allowed to obtain considerable material for the subsequent purification procedure. After a single purification step, the RBD was recovered with a very high purity level (> 95%) assessed by gel densitometry. Throughout RP-HPLC, we could demonstrate the absence of structurally related impurities such as dimers. Because the fluorescence profile is extremely sensitive to perturbations in the local structural environment, it provides a simple and powerful tool to study protein conformational changes. We found that RBD is susceptible to perturbations if it is stressed with a chaotropic agent such as urea, or a strong reducing agent such as DTT, since changes in peak shift or intensity were observed. Besides, when measuring intrinsic protein fluorescence of aromatic amino acids upon excitation at 275 nm, both Trp and Tyr emission spectra can be acquired. When proteins are in their native state, the emission of Tyr is usually quenched by its interaction with the peptide chain or energy transfer to Trp. On the contrary, during protein denaturation, Tyr emission frequently increases, as it can be seen in the modified profile of treated RBD samples.
The SDS-PAGE of RBD treated with PNGase F confirmed the presence of N-glycosylation, which would correspond to glycans attached to the asparagine of the two N-glycosylation consensus sequences (N331 and N343) found in the RBD N-terminal region (Grant et al. 2020). However, the persistence of two bands in RBD after exhaustive digestion with PNGAse F (Fig. 2D, lane 6) suggests the existence of another post-translational modification, most likely, O-glycosylation. Although less is known about the occupancy degree, O-glycosites have been described in T323, S325, T333, S345, and S477 of the receptor-binding domain (Shajahan et al. 2020; Zhang et al. 2021). Indeed, it seems that O-glycosylation at T323 plays a critical role in the virus attachment to the ACE2 epithelial cell receptors (Shajahan et al. 2020) It is largely known that differences in cell lines, culture medium, and culture conditions, among other factors, may affect the glycosylation pattern, so that further glycosylation studies will be necessary to elucidate N- and O-glycan occupancy and structures of RBD produced in this work.
We also demonstrated that the purified RBD sample interacted with the human ACE2 expressed in HEK293 cells and that its conformational state is critical for the SARS-CoV-2 serum reactivity.
Counting with assays with high sensitivity and specificity values is highly desirable. Good sensitivity values correlate with low false-negative rates, that is, low probability that the assay does not detect antibodies even though the person may have them. Particularly, high specificity is required to ensure low false-positive rates, meaning that a small number of people without antibodies may be identified as having antibodies by the assay. High specificity assays may guarantee prediction of good antibody levels. Also, they allow to estimate the need of vaccination and predict optimal timings of booster vaccination (Townsend 2022). The biological basis for false positive is multifactorial, but the influence of the production platform and process-related impurities may be crucial (Klausberger et al. 2021). Klausberger et al. (2021) reported the expression of RBD in HEK, CHO, insect cells, and tobacco plants. They found higher cross-reactivity when RBD was expressed in insect cells in comparison with other expression systems. In agreement with previously reported data (Amanat et al. 2020), the authors considered possible to attribute it to the glycosylation pattern. Besides, Klausberger et al. (2021) found higher yields employing HEK cells as expression system. However, since the expression of RBD in HEK cell lines is transient, variations in RBD dimerization between different production batches were reported. Hence, they produced a truncated RBD version. Likewise, Ayón-Núñez et al. (2022) showed a greater sensitivity value for the eucaryotic-based ELISA (HEK293 cells) in comparison with procaryotic-based assay (Escherichia coli BL21) (90% and 80%, respectively). These results, once again, highlight the importance of the expression system in the quality attributes of the assay, such as sensitivity.
In our work, the use of a recombinant antigen, produced in a stable way and with high quality (high percentage of purity), brings several advantages. It allows us to standardize the assay, avoiding batch-to-batch variations in RBD quality attributes, such as the percentage of dimerization and the percentage of product and process-related impurities, which indirectly improves the assay’s performance. Furthermore, since no infectious viral particles are used, there is no need to work in high-containment biological laboratories.
In order to have a serological assay that could demonstrate possible evidence of acquired immunity, we designed, optimized, and validated an iELISA for the detection of human anti-RBD antibodies. Here, the optimization was achieved applying DoE, one of the main QbD tools. QbD is a systematic approach, approved by the FDA in 2004, which emphasizes product and process understanding based on process control. Analytical methods, like ELISA assays, are considered an integral part of a manufacturing process development. Hence, the application of QbD approach to analytical method development allows to achieve regulatory flexibility, reduce out-of-specification results, and obtain a robust and a cost-effective method (Fukuda et al. 2018). Here, following a multivariate strategy, we identified the optimum conditions within the analyzed design space, reducing the cost and time and increasing the quality of the results in comparison with classical OVAT strategies. At this point, it is important to note that, to our knowledge, this is the first work reporting an ELISA for detecting human anti-RBD antibodies that has been optimized applying QbD concepts, allowing us to define an analytical design space based on the understanding of the developed method. In this sense, working with QbD concepts allowed us to quickly start the method validation stage. It would also facilitate eventual technology transfer.
During the validation process, the optimized iELISA exhibited high sensitivity and specificity values, in line with some previously reported works (or even better) (Ibrahim et al. 2021; Mehdi et al. 2021; Ojeda et al. 2021; Villafañe et al. 2022; Ayón-Núñez et al. 2022; Amanat et al. 2020). However, to make a correct comparison between the performance of different methods, it is necessary to have additional information (such as timing, symptoms), which sometimes is not provided by all the studies. For instance, Mehdi et al. (2021) reported sensitivity values between 55.33 and 92.14%, as a function of the analyzed panel (different times and symptoms). Similarly, Ojeda et al. (2021) reported sensitivity rates ranging from 35 (≤ 7 days of symptoms onset) to 90.4% (after 3 weeks). Ayón-Núñez et al. (2022) reported 100% specificity and 93% sensitivity rates. Amanat et al. 2020 described an ELISA method based on the spike protein expressed in animal cells (Expi293F), evaluating also the RBD domain as coating antigen. They reported good reactivity of the analyzed COVID-19 serum samples and no cross-reactivity with prepandemic serums. However, they did not report sensitivity and specificity values. Our iELISA sensitivity was 94.24 (86.01–98.42%; 95% confidence interval) and the specificity was 95.96% (89.98–98.89%; 95% confidence interval). This high specificity value implies a low false-positive rate and, thus, a lack of cross-reactivity with other human coronaviruses. Furthermore, assay repeatability and intermediate precision rendered acceptable CV% values.
More than 90% of COVID patients produce neutralizing (functional) antibodies. Among them, 90% correspond to RBD-directed antibodies (Konrath et al. 2022). Previous studies demonstrated that RBD-directed antibodies can correlate with neutralizing activity (Byrnes et al. 2020; Ayón-Núñez et al. 2022). Interestingly, since this iELISA can detect antibodies against RBD, it could be proposed as a technique to assess protective immunity. Indeed, to further address the performance of this iELISA, we analyzed 46 plasma samples from donors vaccinated with three types of vaccine platforms. All the evaluated plasma samples exceeded the cut-off value (0.2560), which would indicate, in most of them, the presence of neutralizing antibodies. In recent works, anti-RBD iELISA assays have been used in comparative evolutionary analyses to study vaccine-mediated immunity durability and antibody waning after natural infection, demonstrating to be an excellent correlate of protection against SARS-CoV-2 infection (Townsend 2022). Other study, based on SARS-CoV-2 antibody ELISA and neutralization assay, assessed the immunogenicity and reactogenicity of heterologous vaccination schemes (Pascuale et al. 2022). In this regard, these iELISA results would be used to estimate the likelihood of breakthrough infections over time following vaccination.
The greatest strengths of this iELISA are the good sensitivity and specificity parameters, which were achieved in a short period of time. We attribute these results, on the one hand, to the implementation of a multivariate strategy during the method optimization. Indeed, the implementation of meaningful input (the analyzed factors) and output (the analyzed responses) variables was also crucial. We have obtained an assay with less uncertainty and, thus, less analytical error of the reportable results, which is related to good test parameters such as sensitivity and specificity. Besides, in comparison with traditional method approaches (like OVAT), we have obtained the optimal conditions (an absolute optimum) in the studied domain (the design space). Since during the optimization stage, a polynomial model is obtained, it is possible to know thoroughly the studied system, reducing the out-of-specification results. In contrast, OVAT approach can only find a local maximum, which depends on the initial conditions of the analysis, in a much less cost-effective manner. On the other hand, our results can be ascribed to the use of a high-quality recombinant antigen, produced in a stable manner, contributing to the aforementioned advantages.
We must not fail to mention that, in future assays, to complete the validation process and assess the bias, a larger number of positive (and also negative) samples must be analyzed considering the age, time, and sex sample characteristics and the severity of the disease (mild, moderate, and severe). In this study, such complementary information of the analyzed samples could not be obtained. Of note, since this iELISA is an IgG test, it may probably exhibit a lower performance in terms of sensitivity if a sample panel of earlier stage of the disease (≤ 14 days) is evaluated. Earlier humoral immune response is dominated by IgM and IgA. Indeed, the early neutralizing response is dominated by RBD-IgA-specific antibodies (Sterlin et al. 2021; Zhang et al. 2022).
Moreover, since SARS-CoV-2 mutates over time, genetic variation in the population of circulating viral strains will occur, which may impact the virus’s properties such as the severity of symptoms. The impact of mutations on a test’s performance is influenced by several factors, including the sequence of the variant, the design of the test, and the prevalence of the variant in the population (FDA 2022) In our study, the use of Wuhan-Hu-1 RBD may impact in the sensitivity results, increasing the false-negative rates, particularly if a mutation occurs in the part of the virus genome assessed by the test (in this case, the RBD sequence). However, most vaccine candidates are based on the original Wuhan strain (Hassine 2022); hence, in these cases, the test performance may not be compromised.
To summarize, this iELISA represents a high-throughput technique, requiring a small volume of sample (less than 6 µL), and is simple to perform, reliable, and feasible to be scaled up to satisfy the current demands.
Data availability
All data generated or analyzed during this study are included in this published article (and its supplementary information file).
References
Agrahari R, Mohanty S, Vishwakarma K, Nayak SK, Samantaray D, Mohapatra S (2021) Update vision on COVID-19: structure, immune pathogenesis, treatment and safety assessment. Sensors International 2:100073. https://doi.org/10.1016/j.sintl.2020.100073
Alandijany TA, El-Kafrawy SA, Tolah AM, Sohrab SS, Faizo AA, Hassan AM, Alsubhi TL, Othman NA, Azhar EI (2020) Development and optimization of in-house ELISA for detection of human igg antibody to SARS-CoV-2 full length spike protein. Pathogens 9:1–11. https://doi.org/10.3390/pathogens9100803
Amanat F, Stadlbauer D, Strohmeier S, O Nguyen TH, Chromikova V, McMahon M, Jiang K, Asthagiri Arunkumar G, Jurczyszak D, Polanco J, Bermudez-Gonzalez M, Kleiner G, Aydillo T, Miorin L, Fierer DS, Amarilis Lugo L, Milunka Kojic E, Stoever J, H Liu ST, Cunningham-Rundles C, Felgner PL, Moran T, García-Sastre A, Caplivski D, Cheng AC, Kedzierska K, Vapalahti O, Hepojoki JM, Simon V, Krammer F (2020) A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat Med. https://doi.org/10.1038/s41591-020-0913-5
Ayón-Núñez DA, Cervantes-Torres J, Cabello-Gutiérrez C, Rosales-Mendoza S, Rios-Valencia D, Huerta L, Bobes RJ, Carrero JC, Segura-Velázquez R, Fierro NA, Hernández M, Zúñiga-Ramos J, Gamba G, Cárdenas G, Frías-Jiménez E, Herrera LA, Fragoso G, Sciutto E, Suárez-Güemes F, Laclette JP (2022) An RBD-based diagnostic method useful for the surveillance of protective immunity against SARS-CoV-2 in the population. Diagnostics 12.https://doi.org/10.3390/diagnostics12071629
Bonanni P, Cantón R, Gill D, Halfon P, Liebert UG, Crespo KAN, Martín JJP, Trombetta CM (2021) The role of serology testing to strengthen vaccination initiatives and policies for COVID-19 in Europe. Covid 1:20–38. https://doi.org/10.3390/covid1010004
Byrnes JR, Zhou XX, Lui I, Elledge SK, Glasgow JE, Lim SA, Loudermilk RP, Chiu CY, Wang TT, Wilson MR, Leung KK, Wells JA (2020) Competitive SARS-CoV-2 serology reveals most antibodies targeting the spike receptor-binding domain compete for ACE2 binding. mSphere 5. https://doi.org/10.1128/msphere.00802-20
Castro R, Nobre LS, Eleutério RP, Thomaz M, Pires A, Monteiro SM, Mendes S, Gomes RA, Clemente JJ, Sousa MFQ, Pinto F, Silva AC, Freitas MC, Lemos AR, Akpogheneta O, Kosack L, Bergman ML, Duarte N, Matoso P, Costa J, Bandeiras TM, Gomes-Alves P, Gonçalves CP, Demengeot J, Alves PM (2021) Production of high-quality SARS-CoV-2 antigens: impact of bioprocess and storage on glycosylation, biophysical attributes, and ELISA serologic tests performance. Biotechnol Bioeng 118:2202–2219. https://doi.org/10.1002/bit.27725
Coria LM, Saposnik LM, Pueblas Castro C, Castro EF, Bruno LA, Stone WB, Pérez PS, Darriba ML, Chemes LB, Alcain J, Mazzitelli I, Varese A, Salvatori M, Auguste AJ, Álvarez DE, Pasquevich KA, Cassataro J (2022) A novel bacterial protease inhibitor adjuvant in RBD-based COVID-19 vaccine formulations containing alum increases neutralizing antibodies, specific germinal center B cells and confers protection against SARS-CoV-2 infection in mice. Front Immunol 13:1–17. https://doi.org/10.3389/fimmu.2022.844837
Derringer G, Suich R (1980) Simultaneous optimization of several response variables. J Qual Technol 12:214–219. https://doi.org/10.1080/00224065.1980.11980968
Faizo AA, Alandijany TA, Abbas AT, Sohrab SS, El-Kafrawy SA, Tolah AM, Hassan AM, Azhar EI (2021) A reliable indirect ELISA protocol for detection of human antibodies directed to SARS-CoV-2 Np protein. Diagnostics 11:1–12. https://doi.org/10.3390/diagnostics11050825
FDA (2022) SARS-CoV-2 viral mutations: impact on COVID test Dashboard. https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-viral-mutations-impact-covid-19-tests. Accessed 28 September 2022
Fernandes-Siqueira LO, Ferreira FAP, Sousa BG, Mebus-Antunes NC, Neves-Martins TC, Almeida FCL, Ferreira GC, Salmon D, Wermelinger LS, da Poian AT (2022) On the caveats of a multiplex test for SARS-CoV-2 to detect seroconversion after infection or vaccination. Sci Rep 12. https://doi.org/10.1038/s41598-022-14294-8
Fernández-Barat L, López-Aladid R, Torres A (2020) The value of serology testing to manage SARS-CoV-2 infections. Eur Respir J 56:2–4. https://doi.org/10.1183/13993003.02411-2020
Fukuda IM, Pinto CFF, Moreira CDS, Saviano AM, Lourenço FR (2018) Design of experiments (DoE) applied to pharmaceutical and analytical quality by design (QbD). Braz J Pharm Sci 54:1–16. https://doi.org/10.1590/s2175-97902018000001006
GeurtsvanKessel CH, Okba NMA, Igloi Z, Bogers S, Embregts CWE, Laksono BM, Leijten L, Rokx C, Rijnders B, Rahamat-Langendoen J, van den Akker JPC, van Kampen JJA, van der Eijk AA, van Binnendijk RS, Haagmans B, Koopmans M (2020) An evaluation of COVID-19 serological assays informs future diagnostics and exposure assessment. Nat Commun 11:1–5. https://doi.org/10.1038/s41467-020-17317-y
Goicoechea HC (2018) Chapter 10: experimental design. In: Fingerprinting techniques in food authentication and traceability. pp 187–206
Gourbatsi E, Povey JF, Smales CM (2018) The effect of formulation variables on protein stability and integrity of a model IgG4 monoclonal antibody and translation to formulation of a model ScFv. Biotechnol Lett 40:33–46. https://doi.org/10.1007/s10529-017-2443-x
Grant OC, Montgomery D, Ito K, Woods RJ (2020) Analysis of the SARS - CoV - 2 spike protein glycan shield reveals implications for immune recognition. Sci Rep 1–11. https://doi.org/10.1038/s41598-020-71748-7
Gstöttner C, Zhang T, Resemann A, Ruben S, Pengelley S, Suckau D, Welsink T, Wuhrer M, Domínguez-Vega E (2022) Structural and functional characterization of SARS-CoV-2 RBD domains produced in mammalian cells. 28:56. https://doi.org/10.1021/acs.analchem.1c00893
Harlow E, Lane D (1988) Antibodies A LABORATORY MANUAL. Cold Spring Harbor Laboratory Press, New York, USA, Ch 5
Hassine HI (2022) Covid-19 vaccines and variants of concern: A review. Rev Med Virol 32(4):e2313. https://doi.org/10.1002/rmv.2313
Ibrahim EH, Ghramh HA, Kilany M (2021) Development of in-house ELISAs for the detection of anti-SARS-CoV-2 RBD and N IgG and IgM antibodies in biological samples. J King Saud Univ Sci 33. https://doi.org/10.1016/j.jksus.2021.101439
Klausberger M, Duerkop M, Haslacher H, Wozniak-Knopp G, Cserjan-Puschmann M, Perkmann T, Lingg N, Aguilar PP, Laurent E, de Vos J, Hofner M, Holzer B, Stadler M, Manhart G, Vierlinger K, Egger M, Milchram L, Gludovacz E, Marx N, Köppl C, Tauer C, Beck J, Maresch D, Grünwald-Gruber C, Strobl F, Satzer P, Stadlmayr G, Vavra U, Huber J, Wahrmann M, Eskandary F, Breyer MK, Sieghart D, Quehenberger P, Leitner G, Strassl R, Egger AE, Irsara C, Griesmacher A, Hoermann G, Weiss G, Bellmann-Weiler R, Loeffler-Ragg J, Borth N, Strasser R, Jungbauer A, Hahn R, Mairhofer J, Hartmann B, Binder NB, Striedner G, Mach L, Weinhäusel A, Dieplinger B, Grebien F, Gerner W, Binder CJ, Grabherr R (2021) A comprehensive antigen production and characterisation study for easy-to-implement, specific and quantitative SARS-CoV-2 serotests. EBioMedicine 67. https://doi.org/10.1016/j.ebiom.2021.103348
Konrath KM, Liaw K, Wu Y, Zhu X, Walker SN, Xu Z, Schultheis K, Chokkalingam N, Chawla H, Du J, Tursi NJ, Moore A, Adolf-Bryfogle J, Purwar M, Reuschel EL, Frase D, Sullivan M, Fry B, Maricic I, Andrade VM, Iffland C, Crispin M, Broderick KE, Humeau LMPF, Patel A, Smith TRF, Pallesen J, Weiner DB, Kulp DW (2022) Nucleic acid delivery of immune-focused SARS-CoV-2 nanoparticles drives rapid and potent immunogenicity capable of single-dose protection. Cell Rep 38. https://doi.org/10.1016/j.celrep.2022.110318
Krähling V, Halwe S, Rohde C, Becker D, Berghöfer S, Dahlke C, Eickmann M, Ercanoglu MS, Gieselmann L, Herwig A, Kupke A, Müller H, Klein F, Keller C, Becker S, Neubauer-r P (2021) Development and characterization of an indirect ELISA to detect SARS-CoV-2 spike protein-specific antibodies. J Immunol Methods 490:1–10
Liu G, Rusling JF (2021) COVID-19 Antibody Tests and Their Limitations. ACS Sens 6:593–612. https://doi.org/10.1021/acssensors.0c02621
Malm M, Kuo CC, Barzadd MM, Mebrahtu A, Wistbacka N, Razavi R, Volk AL, Lundqvist M, Kotol D, Tegel H, Hober S, Edfors F, Gräslund T, Chotteau V, Field R, Varley PG, Roth RG, Lewis NE, Hatton D, Rockberg J (2022) Harnessing secretory pathway differences between HEK293 and CHO to rescue production of difficult to express proteins. Metab Eng 72:171–187. https://doi.org/10.1016/j.ymben.2022.03.009
Mehalko J, Drew M, Snead K, Denson JP, Wall V, Taylor T, Sadtler K, Messing S, Gillette W, Esposito D (2021) Improved production of SARS-CoV-2 spike receptor-binding domain (RBD) for serology assays. Protein Expr Purif 179. https://doi.org/10.1016/j.pep.2020.105802
Mehdi F, Chattopadhyay S, Thiruvengadam R, Yadav S, Kumar M, Sinha SK, Goswami S, Kshetrapal P, Wadhwa N, Chandramouli Natchu U, Sopory S, Koundinya Desiraju B, Pandey AK, Das A, Verma N, Sharma N, Sharma P, Bhartia V, Gosain M, Lodha R, Lamminmäki U, Shrivastava T, Bhatnagar S, Batra G (2021) Development of a fast SARS-CoV-2 IgG ELISA, based on receptor-binding domain, and its comparative evaluation using temporally segregated samples from RT-PCR positive individuals. Front Microbiol 11:1–12. https://doi.org/10.3389/fmicb.2020.618097
Mufarrege EF, Antuña S, Etcheverrigaray M, Kratje R, Prieto C (2014) Development of lentiviral vectors for transient and stable protein overexpression in mammalian cells. A new strategy for recombinant human FVIII (rhFVIII) production. Protein Expr Purif 95:50–56. https://doi.org/10.1016/j.pep.2013.11.005
Niwa H, Yamamura K, Miyazaki J (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. https://doi.org/10.1016/0378-1119(91)90434-D
O’Flaherty R, Bergin A, Flampouri E, Mota LM, Obaidi I, Quigley A, Xie Y, Butler M (2020) Mammalian cell culture for production of recombinant proteins: a review of the critical steps in their biomanufacturing. Biotechnol Adv 43:107552. https://doi.org/10.1016/j.biotechadv.2020.107552
Ojeda DS, Lopez Ledesma MMG, Pallarés HM, Costa Navarro GS, Sanchez L, Perazzi B, Villordo SM, Alvarez DE, Echavarria M, Oguntuyo KY, Stevens CS, Lee B, Carradori J, Caramelo JJ, Yanovsky MJ, Gamarnik AV, Long-Ueira Y, Polo ML, Salvatori M, Azzolina S, Ghiglione Y, Salomon H, Quiroga MF, Turk G, Laufer N (2021) Emergency response for evaluating SARS-CoV-2 immune status, seroprevalence and convalescent plasma in Argentina. PLoS Pathog 17. https://doi.org/10.1371/journal.ppat.1009161
Pascuale CA, Varese A, Ojeda DS, Pasinovich ME, Lopez L, Rossi AH, Rodriguez PE, Miglietta EA, Mazzitelli I, di Diego Garcia F, Sanchez L, Rouco SO, Gonzalez Lopez Ledesma MM, Zurano JP, Mazzitelli B, Scruzzi G, Barbero P, Cardozo D, Gallego S, Borda M, Diaz M, Ridao F, Rosales AB, Bhon J, Talia JM, Diangelo ME, Lacaze MA, Aime B, Gutierrez SI, Ercole R, Toro R, Tau L, Delaplace L, Compagnucci MF, Sartori C, Desimone I, Echegoyen C, Velazquez P, Testa C, Hozbor D, Docena G, Laino CH, Kreplak N, Pifano M, Barbas G, Rearte A, Vizzotti C, Castelli JM, Geffner J, Gamarnik AV (2022) Immunogenicity and reactogenicity of heterologous immunization against SARS CoV-2 using Sputnik V, ChAdOx1-S, BBIBP-CorV, Ad5-nCoV, and mRNA-1273. Cell Rep Med 3. https://doi.org/10.1016/j.xcrm.2022.100706
Premkumar L, Segovia-Chumbez B, Jadi R, Martinez DR, Raut R, Markmann AJ, Cornaby C, Bartelt L, Weiss S, Park Y, Edwards CE, Weimer E, Scherer EM, Rouphael N, Edupuganti S, Weiskopf D, Tse LV, Hou YJ, Margolis D, Sette A, Collins MH, Schmitz J, Baric RS, de Silva AM (2020) The receptor-binding domain of the viral spike protein is an immunodominant and highly specific target of antibodies in SARS-CoV-2 patients. Sci Immunol 5:1–10. https://doi.org/10.1126/SCIIMMUNOL.ABC8413
Pulix M, Lukashchuk V, Smith DC, Dickson AJ (2021) Molecular characterization of HEK293 cells as emerging versatile cell factories. Curr Opin Biotechnol 71:18–24. https://doi.org/10.1016/j.copbio.2021.05.001
Rodrigues AF, Soares HR, Guerreiro MR, Alves PM, Coroadinha AS (2015) Viral vaccines and their manufacturing cell substrates: New trends and designs in modern vaccinology. Biotechnol J 10:1329–1344. https://doi.org/10.1002/biot.201400387
Schwarz H, Zhang Y, Zhan C, Malm M, Field R, Turner R, Sellick C, Varley P, Rockberg J, Chotteau V (2020) Small-scale bioreactor supports high density HEK293 cell perfusion culture for the production of recombinant erythropoietin. J Biotechnol 309:44–52. https://doi.org/10.1016/j.jbiotec.2019.12.017
Shajahan A, Supekar NT, Gleinich AS, Azadi P (2020) Deducing the N- And O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 30:981–988. https://doi.org/10.1093/glycob/cwaa042
Stadlbauer D, Amanat F, Chromikova V, Jiang K, Strohmeier S, Arunkumar GA, Tan J, Bhavsar D, Capuano C, Kirkpatrick E, Meade P, Brito RN, Teo C, Mcmahon M, Simon V, Krammer F (2020) SARS-CoV-2 seroconversion in humans: a detailed protocol for a serological assay, antigen production, and test setup. https://doi.org/10.1002/cpmc.100
Sterlin D, Mathian A, Miyara M, Mohr A, Anna F, Claër L, Quentric P, Fadlallah J, Devilliers H, Ghillani P, Gunn C, Hockett R, Mudumba S, Guihot A, Luyt CE, Mayaux J, Beurton A, Fourati S, Bruel T, Schwartz O, Lacorte JM, Yssel H, Parizot C, Dorgham K, Charneau P, Amoura Z, Gorochov G (2021) IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci Transl Med 13(577):eabd2223. https://doi.org/10.1126/scitranslmed.abd2223
Townsend JP (2022) The durability of natural infection and vaccine-induced immunity against future infection by SARS-CoV-2. 1–8. https://doi.org/10.1073/pnas.2204336119/-/DCSupplemental.Published
Villafañe L, Vaulet LG, Viere FM, Klepp LI, Forrellad MA, Bigi MM, Romano MI, Magistrelli G, Fermepin MR, Bigi F (2022) Development and evaluation of a low cost IgG ELISA test based in RBD protein for COVID-19. J Immunol Methods 500:1–5. https://doi.org/10.1016/j.jim.2021.113182
Volkin DB, Sanyal G, Burke CJ, Middaugh CR (2002) Preformulation studies as an essential guide to formulation development and manufacture of protein pharmaceuticals. Pharm Biotechnol 14:1–46
WHO (2022) WHO Coronavirus (COVID-19) Dashboard. https://covid19.who.int/. Accessed 10 October 2022.
Wu CR, Yin WC, Jiang Y, Xu HE (2022) Structure genomics of SARS-CoV-2 and its Omicron variant: drug design templates for COVID-19. Acta Pharmacol Sin 1–13. https://doi.org/10.1038/s41401-021-00851-w
Zhang Y, Zhao W, Mao Y, Chen Y, Zheng S, Cao W, Zhu J, Hu L, Gong M, Cheng J, Yang H (2021) O-Glycosylation landscapes of SARS-CoV-2 spike proteins. Front Chem 9:1–11. https://doi.org/10.3389/fchem.2021.689521
Zhang J, Zhang H, Sun L (2022) Therapeutic antibodies for COVID-19: is a new age of IgM, IgA and bispecific antibodies coming? MAbs 14(1):2031483. https://doi.org/10.1080/19420862.2022.2031483
Zhu J (2012) Mammalian cell protein expression for biopharmaceutical production. Biotechnol Adv 30:1158–1170. https://doi.org/10.1016/j.biotechadv.2011.08.022
Funding
This study was funded by the Agencia Nacional de Promoción de la Investigación, el Desarrollo Productivo y la Innovación (Agencia I + D + i) (IP-COVID-19–224).
Author information
Authors and Affiliations
Contributions
AG, JV, FA, EG, VG, DF, and SA carried out the molecular biology experiments, the generation of recombinant cell lines, and the production of RBD in 1-L bioreactor; MCR, JV, MBT, and AF carried out the purification stage; MCR performed the optimization and validation of the iELISA. MCR, AG, and NC wrote the manuscript. DF and CP revised the manuscript. All the authors approved the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Assays involving human samples were approved and supervised by the Advisory Committee on Ethics and Security of the School of Biochemistry and Biological Sciences, Universidad Nacional del Litoral, according to international guidelines (Council for International Organizations of Medical Sciences (CIOMS), ISBN 92–9036-075–5).
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rodríguez, M.C., Ceaglio, N., Gugliotta, A. et al. Design and optimization of an IgG human ELISA assay reactive to recombinant RBD SARS-CoV-2 protein. Appl Microbiol Biotechnol 106, 7933–7948 (2022). https://doi.org/10.1007/s00253-022-12254-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-022-12254-w