
Abstract
Purpose
Vercirnon is a CCR9 chemokine receptor antagonist being developed for the treatment of Crohn’s disease. As a variety of concomitant medications are often required for the treatment of Crohn’s disease, it is important to characterise the drug interaction profile of vercirnon. To confirm the results of previous in vitro inhibition studies, this study assessed the in vivo effect of vercirnon on the activity of cytochrome P450 enzymes (CYP3A4, CYP2C19 and CYP2C8) and drug transport proteins (BCRP and OATP1B1) using probe substrates.
Methods
This was an open-label, single-sequence, repeat-dose study conducted in 24 healthy adult subjects. On days 1–4, subjects received probe substrates (midazolam, pioglitazone, omeprazole and rosuvastatin; in that order), followed by administration of vercirnon 500 mg twice daily (BID) on days 5–14. On days 11–14, in addition to vercirnon 500 mg BID, subjects also received probe substrates as on days 1–4. Blood samples were collected for pharmacokinetic analysis of probe substrates, vercirnon and two of its metabolites.
Results
Geometric least-squares mean ratios (90 % confidence interval) of area under the concentration-time curve from time zero to infinity for probe administered with vercirnon (test) compared with probe alone (reference) for midazolam, pioglitazone, omeprazole and rosuvastatin were 0.92 (0.85, 0.99), 1.01 (0.95, 1.07), 0.99 (0.76,1.31) and 0.98 (0.88, 1.09), respectively.
Conclusions
Co-administration of probe substrates midazolam, pioglitazone, omeprazole, and rosuvastatin following repeat dosing of vercirnon 500 mg BID demonstrated vercirnon had no clinically significant effect on CYP3A4, CYP2C8, CYP2C19 enzyme activity or BCRP or OATP1B1 transporter activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Vercirnon (formerly CCX282-B or GSK1605786) is a highly potent, orally active, chemokine receptor antagonist whose single target is the G-protein–coupled receptor CCR9, which has been implicated in the aetiology of inflammatory bowel diseases such as Crohn’s disease [1–4]. Crohn’s disease is a chronic, idiopathic, relapsing inflammatory disorder of the gastrointestinal (GI) tract associated with a dysregulated activation of immune cell function. It can affect any portion of the GI tract but most commonly affects the terminal small intestine and colon [5]. Current treatments for subjects with Crohn’s disease consist of corticosteroids, immunosuppressives (e.g., methotrexate, cyclosporine, mercaptopurine and azathioprine), and tumour necrosis factor inhibitors (e.g., infliximab, adalimumab and certolizumab). Although there have been recent advances in the treatment of patients with Crohn’s disease [6], there is still an unmet medical need for new therapies [5, 7]. Currently, patients with Crohn’s disease use multiple prescription and non-prescription medications to treat their inflammatory bowel disease (IBD) symptoms and other systemic and non-IBD disease-related conditions (e.g., diabetes, hypertension, heart disease). Consequently, patients with Crohn’s disease have a potentially higher risk for drug–drug interactions compared with the general population [8].
Following oral administration (250 to 1,000 mg), vercirnon is readily absorbed with a time to maximum concentration (tmax) of 3–4 h and an estimated half-life between 12 and 17 h [9]. Once absorbed, vercirnon is extensively metabolised by multiple oxidative and reductive pathways, including cytochrome P450 (CYP) enzymes such as CYP3A4, CYP2C19 and CYP2B6 (ChemoCentryx, data on file). Vercirnon is passively permeable and unlikely to be a substrate for human p-glycoprotein transporter protein or other transport proteins, and its metabolites are highly protein bound (>90 %) with low uptake of drug-related material into tissues (ChemoCentryx, data on file). Based on these observations, the potential for other drugs to have a clinically-significant effect on vercirnon was predicted to be low.
No clinical studies have been performed to evaluate the potential for vercirnon to affect the pharmacokinetics (PK) of other drugs. Due to the high plasma protein binding for vercirnon and its metabolites, and the relatively low anticipated liver concentrations estimated from likely therapeutic plasma concentrations (maximum observed concentration [Cmax] range 3–5 μM for vercirnon and <0.5 μM for metabolites; ChemoCentryx, data on file), the potential for vercirnon to significantly affect the clearance of other drugs utilising CYP enzymes as a primary clearance pathway was considered to be low. However, the estimated [I]/Ki ratio, where [I] represents the mean steady-state Cmax value for total drug (bound plus unbound) following administration of the highest proposed clinical dose of vercirnon, was between 0.1 and 1.0 for cytochrome P450 enzymes CYP3A4, CYP2C19, and CYP2C8 activity. This indicated a “possible” interaction requiring a follow-up clinical evaluation [10]. In addition, in vitro studies showed that vercirnon can inhibit breast cancer resistant protein (BCRP) and organic anion transporter (OAT) proteins, specifically OAT1B1, OAT1 and OAT3 (ChemoCentryx and GlaxoSmithKline, data on file).
It is important to confirm these in vitro findings and characterise the drug interaction profile of vercirnon in humans to support late-phase clinical trials, as patients with Crohn’s disease are often prescribed multiple medications, such as immunosuppressants, GI anti-inflammatory agents, antibiotics and pain relievers. This study investigated the potential effect of steady-state vercirnon on CYP3A4, CYP2C19 and CYP2C8 activity, plus BCRP and OATP1B1 transporter activity, by evaluating the effect of vercirnon co-administration on the PK of well-established probe substrates for each enzyme/transporter [11]. As a secondary objective, the systemic exposure of two metabolites: GSK2635622 and GSK2656694, relative to the parent drug, vercirnon, were assessed following single and repeat dosing of vercirnon. In addition, exploratory pharmacogenetic (PGx) evaluations were conducted to investigate the relationship between genetic variants in drug metabolism and transporter genes, and inter-individual variability in PK parameters.
Methods
Subjects
A total of 24 subjects were enrolled to ensure that at least 16 subjects would complete the study. Subjects were eligible to participate if they were a healthy adult male or female, of non-childbearing potential, 18–55 years of age with body weight ≥60 kg and body mass index (BMI) within the 19–30 kg/m2 range. Subjects were excluded from the study if they were pregnant, smoked, or had liver disease, active or latent tuberculosis, coeliac disease or a positive serologic test for tissue transglutaminase (tTG), hepatitis B, hepatitis C, or human immunodeficiency virus (HIV). Subjects were also excluded if they had drug or alcohol problems, blood pressure abnormalities, mental/legal incapacities, sensitivities, medical conditions judged by the principal investigator to be unsuitable for the study, or if they had consumed red wine, citrus fruits, fruit juices, vegetables from the mustard green family or charbroiled meats within 7 days prior to the first dose of study medication. No alcohol, caffeine, or concomitant medications were allowed while in the clinic. All subjects provided written informed consent before any study-specific procedures were performed.
The study was conducted in accordance with good clinical practice and all applicable regulatory requirements under the Declaration of Helsinki at a single site: Buffalo Clinical Research Center, Buffalo, NY, USA. A local Institutional Review Board reviewed and approved the study.
Study design and treatments
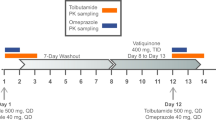
This was an open-label, single-sequence, repeat-dose study (CCX114671; ClinicalTrials.gov identifier NCT01489943) designed to evaluate the effect of repeat doses of vercirnon on the PK of probe substrates of three CYP enzymes and two transporter proteins in healthy subjects. The probes used to investigate the enzyme and transporter activity were: midazolam for CYP3A4, pioglitazone for CYP2C8, omeprazole for CYP2C19, and rosuvastatin for BCRP and OATP1B1. All probe substrates were administered orally and at doses that would provide adequate concentrations of parent and/or metabolite for the assessment of potential interactions with vercirnon. The probe compounds were administered in the morning on the study days outlined in Table 1. Vercirnon 500 mg was administered twice a day (BID), after a meal, on days 5–14. Eligible subjects were admitted to the clinical unit on day -1 (day before first dose) and remained in-house until day 15. On days 1, 2, 3 and 4 they received single doses of the probe compounds: midazolam 3 mg oral syrup, pioglitazone 15 mg tablet, omeprazole 40 mg capsule and rosuvastatin 10 mg tablet, respectively. This was followed by vercirnon 2 x 250-mg capsules BID on days 5–10. On days 11, 12, 13 and 14, in addition to vercirnon 500 mg BID, subjects also received the probe compound in the same order as days 1–4.
For each morning dose, subjects were required to fast overnight. Subjects were served a moderate fat breakfast (i.e., fed state) consisting of approximately 28 % fat and 700 cal. Thirty minutes after breakfast was served, study medication was administered with 240 mL of water. For each evening dose of vercirnon, subjects were also served a standard meal 30 min prior to dosing. Serial blood samples were collected over a 24-h period following the administration of all probe substrates; except for rosuvastatin, where samples were collected over 48 h. Full details of sample collection and probe substrate analysis (including validation, precision and accuracy ranges) are provided in Online Resource 1. Systemic levels of the probe substrates when co-administered with vercirnon were compared with systemic levels when the probes were administered alone. On study days 5–10, single trough concentrations were collected immediately prior to dosing. Additionally, a subset of subjects volunteered to have serial blood samples collected over 24 h on study days 5 and 10 for analysis of vercirnon and its metabolites, GSK2635622 and GSK2656694. All subjects returned to the unit for a follow-up visit within 7–10 days after the last dose of study medication before being discharged from the study. The total duration of a subject’s participation in the study, including screening (∼30 days), treatment and the follow-up visit was approximately 54 days.
Sample size considerations
The potential effect of vercirnon on the probe substrates evaluated in this study was assessed by comparing the ratio of area under the concentration curve from zero to infinity (AUC0–∞) of the probe substrate in combination with vercirnon (test) to the AUC0–∞ of the probe substrate alone (reference). The precision for the comparison of interest was estimated, using a sample size of 16 subjects and based on the largest average estimate of within-subject variability (%CVw) for the CYP450 substrates to be studied (28.0 % for rosuvastatin AUC0–∞; GlaxoSmithKline, data on file). Point estimates and corresponding 90 % confidence intervals (CIs) were constructed for the differences between the mean of the test and reference treatments (the comparison of interest). The estimated precision is no more than 19 % of the observed point estimate, where precision is expressed as the half-width of the 90 % CI. For a point estimate of one, the corresponding 90 % CI for the ratio of geometric means would be 0.84–1.19. This calculation was based on the loge-scale and a two-tailed procedure with a type I error rate of 10 %. A total of 24 subjects were enrolled to ensure 16 patients would complete the study.
A sensitivity analysis was to be conducted in the event that the %CVw estimates were greater than expected. For the %CVw in the upper range of 35 %, it was estimated that the precision for the comparisons of interest would be no more than 23 % of the observed point estimate (i.e., the 90 % CI for the ratio of geometric means would be 0.81–1.23, with a point estimate of one). This calculation was based on the loge-scale and a two-tailed procedure with a Type I error rate of 10 %.
Study population
The ‘safety population’ consisted of all subjects who received at least one dose of study medication. The ‘PK population’ included subjects in the ‘safety population’ with PK samples adequate for calculation of PK parameters. The ‘day 5 and day 10 population’ excluded subjects from the PK population who had multiple PK samples collected on day 5 and day 10. The ‘PGx population’ excluded subjects who were predicted to be poor metabolisers or have potentially reduced transporter activity based on genotyping.
Pharmacokinetic analysis
Plasma concentration time data for all probe substrates, vercirnon and metabolites were analysed by non-compartmental methods using WinNonlin® v5.3 (PharSight, a Certara Company, St Louis, MO). From the concentration time data for each probe compound, the AUC0–∞, the AUC time curve from time zero (pre-dose) to last time of quantifiable concentration (AUC0–t), Cmax, time of occurrence of Cmax (tmax), and apparent terminal phase half-life (t1/2) (where data were available) were determined following administration with and without vercirnon.
For vercirnon and its two metabolites, the AUC time curve over the dosing interval (AUC0–τ), Cmax and tmax were determined in the subset of six subjects who had serial PK sampling on days 5 and 10. Vercirnon accumulation following repeat dosing was determined by calculating the accumulation ratio (Ro) for AUC0–τ and comparing the Cmax for day 10 with day 5. The ratio of metabolite AUC0–τ to vercirnon AUC0–τ on day 5 and day 10 was calculated after converting the AUC0–τ values to appropriate molar units.
Statistical analysis
Probe assessment
The potential effect of vercirnon on the probe substrates evaluated in this study was assessed by comparing the ratio of AUC0–∞ of the probe substrate with and without concomitant vercirnon. The primary endpoint for all probe substrates (except omeprazole) was the ratio of AUC0–∞ for each of the probe substrates when administered with and without concomitant vercirnon. Due to the high inter- and intra-subject variability associated with omeprazole PK [12], its metabolite (5-OH-omeprazole) was also quantified in plasma and the ratio of omeprazole metabolite to parent was calculated to normalise for the variability associated with the absorption and metabolism of omeprazole. The statistical endpoint for omeprazole was calculated from the AUC0–∞ ratio of metabolite:parent, both with and without concomitant vercirnon. Following loge-transformation, the primary endpoints were separately analysed using a mixed-effects model. Treatment was fitted as a fixed effect and subject as a random effect. Point estimates and their associated 90 % CI were constructed for the ratios, by back transforming the respective difference between the treatment least squares (LS) means and the associated 90 % CI.
Vercirnon steady-state analysis
Trough concentration (Cτ) samples collected from all 24 subjects on study days 6–11 were used to assess attainment of steady-state. Individual plasma vercirnon pre-dose concentrations were obtained and the mean (+ standard deviation [SD]) and median (range) were plotted and summarised by day. A mixed-effects model with subject as a random effect and day as a fixed effect was performed on the loge-transformed Cτ data. Day was treated as a continuous variable in the model. Achievement of plasma vercirnon steady state was assessed by calculating the 90 % CI of the slope of the linear regression of pre-dose Cτ versus day. To claim steady state was reached, the slope estimate needed to be close to zero or the 90 % CI for the slope estimate needed to include zero.
Safety
Safety and tolerability parameters were assessed by the measurement of vital signs, cardiac monitoring, adverse event (AE) assessments and clinical laboratory tests. The two vercirnon metabolites (GSK2635622 and GSK2656694) were assayed and the molar ratio of each metabolite to vercirnon (parent) concentrations were determined.
Pharmacogenetic analyses
To account for potential inter-patient variability as a result of genetic polymorphisms, exploratory PGx evaluations were also conducted. Exploratory analyses included PK parameters in a sub-population of subjects who had no common genetic variations in the CYP enzymes or BCRP/OATP1B1 transporters that could negatively impact their activity (the PGx population). The ‘Affymetrix DMET-Plus’ genotyping array [13] was used to genotype common genetic variants in CYP3A4/3A5, CYP2C8, CYP2C19, SLCO1B1 (OATP1B1) and ABCG2 (BCRP). Subjects carrying genetic variants which predicted poor metabolism or reduced activity for the respective CYP enzymes or transporters were excluded from the relevant exploratory PGx analysis, thereby creating sub-populations with fewer confounding factors.
Results
Subjects
A total of 24 subjects (23 males: 1 female) enrolled and completed the study. Subjects had a mean (±SD) age of 33 ± 10.6 years (range 19–54 years), weighed 78.7 ± 12.8 kg (range 61.0–109.9 kg) and had a mean (±SD) BMI of 25.68 ± 2.77 kg/m2 (range 20.1–30.2 kg/m2). Sixteen subjects (67 %) were Caucasian or of European heritage and 8 (33 %) were African-American or of African heritage.
All 24 (100 %) subjects enrolled were included in the ‘safety population’ and the ‘PK population’. The ‘day 5 and day 10 population’ consisted of six subjects whilst the ‘BCRP PGx’ and ‘OATP1B1 PGx’ populations consisted of 19 and 22 subjects, respectively.
Pharmacokinetics
Probe and treatment comparisons
The plasma PK parameters for probe substrates midazolam, pioglitazone, omeprazole, 5-OH-omeprazole and rosuvastatin administered with and without concomitant vercirnon are summarised in Table 2. PK parameters for each of the probe substrates were similar irrespective of whether they were administered alone or in combination with vercirnon. AUC0–∞ variability for each of the probe substrates are presented in Table 3. In the case of omeprazole, the % CVw for the AUC0–∞ treatment comparison was 54.3 % (Table 3). The %CVw was greater than the 28 % used to estimate sample size and the precision of the comparison of interest. However, using the planned ratio of the 5-OH-omeprazole metabolite AUC0–∞ to omeprazole AUC0–∞, the %CVw was reduced to 13.7 %.
The %CVw for AUC0–∞ for midazolam, pioglitazone, rosuvastatin and 5-OH-omeprazole:omeprazole, (with and without vercirnon) ranged from 11.6 % to 21.7 %. The 90 % CI of the geometric LS mean ratios for midazolam, pioglitazone, rosuvastatin, and 5-OH-omeprazole:omeprazole all fell within the 0.80–1.25 range of standard bioequivalence (Table 3).
Vercirnon and metabolites—accumulation and steady-state assessment
A summary of the derived vercirnon PK parameters on study day 5 (day 1 of vercirnon dosing) and study day 10 (day 6 of vercirnon repeat dosing) for the 6 subjects who participated in the serial PK sampling are presented in Table 4. The average accumulation for vercirnon in terms of AUC0–τ and Cmax on day 10, compared with day 5, was 14 % and 3 %, respectively. Concentration-time profiles for the metabolites GSK2656694 and GSK2635622 paralleled that of vercirnon (Fig. 1) and appear (by visual inspection) to accumulate and reach steady-state after 2 days of dosing (∼48 h). The statistical assessment of vercirnon steady-state, following daily dosing of vercirnon 500 mg twice daily for 6 days, confirmed that plasma concentrations of vercirnon achieved steady-state (90 % CI of the slope estimates included zero) by the end of the second day of dosing. When vercirnon was at steady-state (day 10), the mean AUC0–τ for the metabolites GSK2635622 and GSK2656694 averaged 1.1 % and 2.6 % of parent AUC0–τ, respectively. Based on the data from the six subjects in this study, neither metabolite would be predicted to exceed 10 % of parent exposure.

Mean (95 % CI) plasma concentrations of vercirnon and metabolites GSK2635622 and GSK2656694 following vercirnon 500 mg BID for 6 days (n = 6)
Safety
Vercirnon, along with the four probe substrates, was well tolerated in the 24 subjects who completed all treatments over the course of 14 days. There were no deaths, non-fatal serious AEs, early withdrawals due to AEs or pregnancies. A total of 43 post-treatment AEs were reported during the study. The most frequent AE was somnolence, which was reported by 13 subjects on 19 occasions. In all cases, subjects were being treated with either midazolam alone or a combination of midazolam and vercirnon. Eight gastrointestinal AEs (dyspepsia [n = 3], nausea [n = 2], and one each of abdominal pain, diarrhoea and gastro-oesophageal reflux) were reported by seven subjects. Three of these AEs were classified as potentially drug-related by the investigator, though only two (abdominal pain, dyspepsia) were reported during treatment with vercirnon alone. Other non-gastrointestinal AEs that occurred in more than one subject were headache (n = 4), dizziness (n = 2), and abnormal dreams (n = 2). All AEs were classified as mild in intensity with the exception of two clinical chemistry laboratory AEs in one subject, which were judged by the investigator to be of severe intensity, but unrelated to study treatment. One subject who had normal aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels throughout the study and at discharge was also recorded as having elevated ALT and AST values at an unscheduled visit (when the subject returned to the clinic to screen for another clinical study). This was 16 days after the last dose of vercirnon and rosuvastatin, when drug concentrations would be negligible. Additional information provided by the investigator confirmed that the subject remained well and that the transaminase levels had returned to normal over a few weeks.
All AEs resolved by the end of the study and no other clinically significant laboratory findings, haematology, vital signs, or electrocardiogram (ECG) changes were observed.
Pharmacogenetics
PK analyses were performed for the BCRP and OATP1B1 transporter PGx sub-populations only, as no subjects were predicted to be poor metabolisers based on their CYP3A4/3A5, CYP2C8 or CYP2C19 genotype. An evaluation of PK endpoints in the BCRP (n = 19) and OATP1B1 (n = 22) PGx populations revealed no significant difference in PK outcomes compared with the overall study population. All geometric mean ratios were between 0.98 and 1.01 and the 90 % CI were between 0.86 and 1.13.
Discussion
Assessing potential drug–drug interactions is a key aspect in the development of any pharmaceutical treatment, especially in the assessment of safety and effectiveness. The objective of drug–drug interaction studies is to determine the potential for interactions and whether these may require dosage adjustments or additional therapeutic monitoring to maintain treatment efficacy and/or mitigate risk. Preclinical studies identified a potential interaction between vercirnon and CYP3A4, CYP2C19, and CYP2C8 activity with [I]/Ki ratios between 0.1 and 1.0, which would require a follow-up clinical evaluation in line with the recommendations in the US Food and Drug Administration Guidance for Industry: Drug Interaction Studies [10], as well a potential interaction with the transporter proteins BCRP, OAT1B1, OAT1 and OAT3 (ChemoCentryx and GlaxoSmithKline, data on file). Since the in vitro results were similar, these studies did not help to prioritise the evaluation of clinical drug interactions. Given the number of potential interactions, this study was designed to utilise probe substrates to evaluate the effect of vercirnon on the activity of all the CYP enzymes and transporter proteins of interest in healthy adult subjects.
Ideally, selected probe substrates should be the most sensitive and specific substrates for the CYP enzymes or transport proteins under investigation. Midazolam and omeprazole are recognised as sensitive probe substrates for CYP3A4 and CYP2C19, respectively: their AUC0–∞ values would be expected to increase by 5-fold when co-administered with a known CYP3A4 or CYP2C19 inhibitor. Pioglitazone was used as the probe substrate for CYP2C8 instead of the more sensitive probe substrate repaglinide, because it has a lower potential for hypoglycaemia in healthy non-diabetic subjects, and because it is also a substrate for OATP1B1; in vitro studies have shown that vercirnon has the potential to inhibit OATP1B1 transport proteins. CYP2C8 and CYP3A4 are the main enzymes catalysing the transformation of pioglitazone. Co-administration of pioglitazone with gemfibrozil (Ki = 69–75 μM), a strong inhibitor of CYP2C8, significantly increases pioglitazone AUC by 3.4-fold [14]. Co-administration with rifampin, a potent inducer of CYP2C8, decreases the exposure (AUC) of pioglitazone by 54 % [15]. However, co-administration of pioglitazone with ketoconazole, a strong inhibitor of CYP3A4, only increases pioglitazone by 34 %. For these reasons, pioglitazone was considered a suitable probe substrate for CYP2C8.
Drug transport proteins affect the concentration of drugs and metabolites in various tissues and can have a significant effect on the safety profile of a drug. Rosuvastatin, an HMG-CoA reductase inhibitor indicated for the treatment of hypercholesterolaemia, is recognised as a probe substrate for multiple transporters: OATP1B1, OATP1B3, OAT3 and BCRP [11]. Rosuvastatin is actively transported into hepatocytes by OATP1B1, resulting in a high first-pass effect and a low bioavailability (∼20 %). Co-administration of cyclosporin, an inhibitor of OATP1B1, increased rosuvastatin AUC by 7-fold [16]. A recent in vitro study suggested that the BCRP efflux transporter may also play a role in rosuvastatin disposition [17]. While rosuvastatin may not be able to identify which transporter protein vercirnon may affect, a negative result would rule out drug interaction due to several transporters and confirm rosuvastatin as an anti-cholesterol agent that may be co-administered with vercirnon.
Overall, administration of midazolam, pioglitazone, omeprazole, or rosuvastatin following repeat dosing of vercirnon had no significant effect on the systemic exposure (AUC0–∞) of each probe. This indicates that vercirnon exerted no substantial clinical effect on the activity of CYP enzymes CYP3A4, CYP2C8, CYP2C19 or on BCRP and OAT1B1 transporter activity.
In the case of omeprazole, the %CVw for the AUC0–∞ treatment comparison was particularly high (54.3 %). This could be an outlier effect, as one subject recorded the lowest exposure when receiving omeprazole alone, followed by one of the highest exposures when he received omeprazole in combination with vercirnon. Additional statistical analysis, excluding this subject from the omeprazole analysis, reduced the %CVw for omeprazole to 27.8 %. However, excluding this subject did not have a substantial impact on the overall findings. While safety and tolerability data were collected in this study, it must be noted that the study was not powered to assess these outcomes, and any interpretation should be made with caution. Nevertheless, the results of this study suggest that short-term exposure to vercirnon appears to have a favourable safety and tolerability profile. This requires confirmation in future studies. In addition, although there is a low predicted potential for vercirnon to interact with other drugs, or vice versa, this study did not directly assess the interaction of vercirnon with current treatments for Crohn’s disease, such as corticosteroids, immunosuppressives, and tumour necrosis factor inhibitors. Vercirnon is a distinct class of compound to these treatments, and so the potential for combination therapy warrants investigation in future studies.
In conclusion, the results of this study confirm and extend those from in vitro studies, demonstrating that vercirnon is unlikely to affect the clearance of other drugs metabolised by CYP3A4, CYP2C8 and CYP2C19 enzymes, or affect the transport activity of BCRP or OATP1B1. This indicates that there is a low risk of drug–drug interactions with repeat doses of vercirnon, supporting its use in Phase III clinical trials.
References
Wurbel MA, Philippe JM, Nguyen C, Victorero G, Freeman T, Wooding P et al (2000) The chemokine TECK is expressed by thymic and intestinal epithelial cells and attracts double- and single-positive thymocytes expressing the TECK receptor CCR9. Eur J Immunol 30:262–271
Papadakis KA, Prehn J, Nelson V, Cheng L, Binder SW, Ponath PD, Andrew DP, Targan SR (2000) The role of thymus-expressed chemokine and its receptor CCR9 on lymphocytes in the regional specialization of the mucosal immune system. J Immunol 165:5069–5076
Zabel BA, Agace WW, Campbell JJ, Heath HM, Parent D, Roberts AI et al (1999) Human G protein-coupled receptor GPR-9-6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. J Exp Med 190:1241–1256
Walters MJ, Wang Y, Lai N, Baumgart T, Zhao BN, Dairaghi DJ et al (2010) Characterization of CCX282-B, an orally bioavailable antagonist of the CCR9 chemokine receptor, for treatment of inflammatory bowel disease. J Pharmacol Exp Ther 335:61–69
Abraham C, Cho JH (2009) Inflammatory bowel disease. N Engl J Med 361:2066–2078
Keshav S, Vanasek T, Niv Y, Petryka R, Howaldt S, Bafutto M et al (2013) A randomized controlled trial of the efficacy and safety of CCX282-B, an orally-administered blocker of chemokine receptor CCR9, for patients with Crohn’s disease. PLoS One 8:e60094
Marsal J, Agace WW (2012) Targeting T-cell migration in inflammatory bowel disease. J Intern Med 272:411–429
Buckley JP, Kappelman M, Allen JK, Van Meter SA, Cook S (2013) The burden of comedication among inflammatory bowel disease patients. Gastroenterology 144(Supp 1):S-388
Haberer LJ, Hacquoil K, Ino H, Sakamoto T, Kanemoto N, McSherry I et al (2013) Single-dose pharmacokinetics of the CCR9 receptor antagonist vercirnon in healthy US and Japanese subjects. Clin Pharmacol Drug Dev. doi:10.1002/cpdd.41
US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) (2006) Guidance for Industry: Drug Interaction Studies—Draft Guidance. Study Design, Data Analysis, and Implications for Dosing and Labeling
US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) (2012) Guidance for Industry: Drug Interaction Studies—Draft Guidance. Study Design, Data Analysis, and Implications for Dosing and Labeling. Accessed September 2013 at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf.
Lawson EB, Wu JC, Baldwin RM, Ingelman-Sundberg M, Rosenborg S, Yim DS et al (2012) Omeprazole limited sampling strategies to predict area under the concentration-time curve ratios: implications for cytochrome P450 2C19 and 3A phenotyping. Eur J Clin Pharmacol 68:407–413
Deeken J (2009) The Affymetrix DMET platform and pharmacogenetics in drug development. Curr Opin Mol Ther 11:260–268
Deng LJ, Wang F, Li HD (2005) Effect of gemfibrozil on the pharmacokinetics of pioglitazone. Eur J Clin Pharmacol 61:831–836
Jaakkola T, Backman JT, Neuvonen M, Laitila J, Neuvonen PJ (2005) Effect of rifampicin on the pharmacokinetics of pioglitazone. Br J Clin Pharmacol 61:70–78
Simonson SG, Raza A, Martin PD, Mitchell PD, Jarcho JA, Brown CD et al (2004) Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther 76:167–177
Huang L, Wang Y, Grimm S (2006) ATP-dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug Metab Dispos 34:738–742
Acknowledgments
The authors wish to thank Robert Blum and the staff at Buffalo Clinical Research Center (BCRC), Buffalo, NY, US for their support with the study; Beth Harris and Louise Drewett for assistance in analysing genetic outcomes; and Gary Bowers, Grant Generaux, and Joe Polli for scientific consultation. Editorial support was provided by Karen Yee and Stuart Wakelin at Fishawack Scientific Communications Ltd.
Funding disclosure
This study was funded by GlaxoSmithKline. Statistical analysis, provided by PHASTAR, Chiswick, UK, and editorial support, provided by Fishawack Scientific Communications Ltd., were funded by GlaxoSmithKline.
Declaration of conflict of interest
Lynda J Haberer, Iain McSherry, and Linda McCarthy are GlaxoSmithKline employees and hold GlaxoSmithKline stock and stock options. Anna Cargill is an employee of PHASTAR, Chiswick, UK (funded by GlaxoSmithKline to provide statistical analysis).
Author information
Authors and Affiliations
Corresponding author
Additional information
Contributions of authors statement
All authors were involved in the conception and design of the study as well as in the analysis and interpretation of data. Linda McCarthy was also involved in acquisition of data. All authors contributed to drafting the manuscript, revised it critically for important intellectual content and provided final approval for the final version to be published.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 429 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Haberer, L.J., McSherry, I., Cargill, A. et al. Effects of vercirnon on the activity of CYP3A4, CYP2C19 and CYP2C8 enzymes and BCRP and OATP1B1 transporters using probe substrates. Eur J Clin Pharmacol 70, 37–45 (2014). https://doi.org/10.1007/s00228-013-1592-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-013-1592-7