
Abstract
A two-dimensional chromatographic method with mass spectrometric detection has been developed for identification of small, hydrophilic angiotensin I-inhibiting peptides in enzymatically hydrolysed milk proteins. The method involves the further separation of the poorly retained hydrophilic fraction from a standard C18 reversed-phase column on a hydrophilic interaction liquid chromatography (HILIC) column. The latter column is specifically designed for the separation of hydrophilic compounds. Narrow fractions collected from the HILIC column were analysed for their angiotensin I-converting enzyme (ACE) inhibiting potential in an at-line assay. Fractions showing significant inhibition of ACE were analysed by LC–MS for structure elucidation. With this method the main peptides responsible for ACE-inhibition in the hydrophilic part of a milk hydrolysate could be determined. The ACE-inhibiting peptides RP, AP, VK, EK, and EW explained more than 85% of ACE-inhibition by the hydrophilic fraction.
Similar content being viewed by others


Introduction
Hypertension is one of the major health problems in the western world. A variety of food ingredients that help to reduce blood pressure have been described. Particularly attractive are hydrolysed proteins from fermented milk containing specific peptides that can inhibit the angiotensin I-converting enzyme (ACE), an enzyme involved in blood-pressure regulation [1–3]. For the development of optimized anti-hypertensive food products, it is crucial to know which specific peptide(s) in the complex protein hydrolysate is (are) responsible for the ACE-inhibition.
Identification of active ingredients in complex mixtures is generally done using assay-guided repeated fractionation. For the specific case of ACE-inhibiting peptides in milk-protein hydrolysates often only part of the total activity of the product can be assigned to specific peptides [4]. A significant part of the remaining activity is present in fractions that are poorly retained by the standard reversed-phase columns used in the fractionation procedure. Nakamura et al. [1] for example could only explain part of the activity of a casein hydrolysate by the tri-peptides VPP and IPP eluting in one of the later fractions. The remaining activity was present in the unretained fraction. In our laboratory similar samples were seen to have over 30% of the total activity in the unretained fraction. Identification of the oligopeptides in this fraction was not further pursued but would have been extremely difficult because the fraction contains far too many compounds for unambiguous activity assignment. Moreover, there are also too many interfering compounds present in the first-eluting fractions for reliable MS identification. Similar situations are also described by other authors [5–7]. In a previous investigation we presented a new at-line method for the identification of ACE-inhibiting (oligo)peptides in hydrolysed caseinate [4]. Approximately 70% of the activity of these samples was caused by peptides nicely retained by the two reversed-phase columns used in the fractionation. Twenty percent of the activity was present in the hydrophilic fraction. The remaining 10% was caused by a large number of peptides with low ACE-inhibiting activity. Identification of the peptides in the hydrophilic fraction was impossible due to the large number of peptides present and the presence of co-eluting compounds, such as salts and sugars, that resulted in severe suppression of ionisation. Two reversed-phase columns were used in an at-line 2D set-up to provide improved resolution of the very complex mixture. Unfortunately, the highly hydrophilic peptides escaped identification since they were unretained on both columns. To identify the peptides responsible for the ACE inhibition of the unretained hydrophilic fraction improved chromatographic retention and separation is required. Hydrophilic interaction chromatography (HILIC) could be an attractive way of achieving this.
The use of HILIC for separation of hydrophilic substances such as proteins, peptides, and nucleic acids was first described by Alpert in 1990 [8]. Since then HILIC has been used for the separation of many different substances, for example denaturants [9], polar oligomers [10], and pharmaceutical ingredients [11]. Many publications can be found describing the increased retention of hydrophilic peptides on HILIC columns in comparison with reversed-phase columns. A detailed review is published by Yoshida [12]. Literature describing the application of reversed-phase separation in combination with HILIC chromatography is scarce. A notable exception is the compositional analysis of an enzymatic protein hydrolysate obtained from acid-deaminated wheat gluten by Schlichtherie-Cerny [13]. The hydrolysate was first fractionated by GPC. The fraction corresponding to a molecular weight range of 100–750Da was then further fractionated by RPLC. Finally, the peptides in the unretained peak from the RPLC column were separated and characterised by HILIC–API-MS. Using this method ten amino acids, eight di-peptides and one tri-peptide, were identified in this fraction.
In this paper we describe the development of a method for the identification of ACE inhibiting peptides in the poorly retained fractions of a reversed-phase fractionation of an enzymatically hydrolysed milk protein. The possibility of improving chromatographic resolution by using a HILIC separation as a second-dimension separation after the first reversed-phase isolation is studied. The aim of the new method is to extend the applicability of a previously developed system for the identification of ACE-inhibiting peptides in complex peptide mixtures such as protein hydrolysates [4]. The applicability of the method is demonstrated by the identification of hydrophilic ACE-inhibiting peptides in an enzymatically hydrolysed milk protein.
Experimental
Chemicals
The model peptides VPP, IPP, and LPP (purity >98%) were purchased from Bachum (Dübendorf, Switzerland). All other peptides were synthesised by JPT Peptide Technologies (Berlin, Germany). For HPLC analysis, acetonitrile, formic acid, and trifluoroacetic acid were purchased from Merck (Amsterdam, The Netherlands). Aqueous ammonia (Merck) was used for neutralization of the HPLC fractions. For instrument tuning PEG 300, PEG 600, and PEG 1000 were purchased from Sigma Chemicals (St Louis, MO, USA). Ammonium acetate was obtained from Merck. For the at-line assay angiotensin I-converting-enzyme (ACE) and hippuryl–histidyl–leucine (HHL) were purchased from Sigma–Aldrich Chemie (Zwijndrecht, The Netherlands). PBS buffer was purchased from Gibco (Paisley, UK). The hydrolysed milk powder was an enzymatically hydrolysed milk protein obtained from Calpis (Tokyo, Japan).
Instrumentation
All peptide separations and identifications were performed with a Waters Alliance 2795 HT HPLC coupled to a Micromass QTOF-Ultima hybrid time-of-flight mass spectrometer equipped with a lock spray option for accurate mass determination (Waters, Almere, The Netherlands). Fractions were collected in polypropylene 300-μL 96-well plates (Nunc, Roskilde, Denmark) using a Mark IV fraction collector also from Waters. Solvent evaporation was performed using an Ultravap 96-well evaporation device (Porvair, Shepperton, UK). Quantification of HHL and HL in the at-line assay was performed on the QTOF instrument described above.
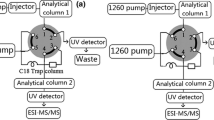
Two-dimensional analytical separation
The first-dimension reversed-phase separation was performed on two serially connected 150 × 2.1mm Inertsil 5 ODS3 columns, particle size 5μm, operating in the gradient mode (Varian, Middelburg, The Netherlands). The guard column was a 20 × 3.9mm SymmetryShield RP8 column packed with 5-μm particles (Waters, Etten-Leur, The Netherlands). Mobile phase A consisted of 0.1% TFA in Milli-Q water. Mobile phase B consisted of 0.1% TFA in acetonitrile. The initial eluent composition was 100% A. After a 5min hold a linear gradient was started to 5% B in 15min, followed by a linear gradient to 70% B in 75min. Finally, mobile phase composition was programmed to 99% B in 1min and kept at 99% B for another 4min. At the end of the gradient the eluent composition was reduced to 100% A in 6min and the system was allowed to re-equilibrate for 14min. The total run time was 120min. The eluent flow rate was 0.2mL min−1 and the column temperature was 60°C. In-house experiments revealed that at this temperature isomeric peptides such as IPP and LPP are separated. UV detection was performed at 215nm and 254nm. Fractions of 200μL were collected in 96-well plates. After neutralization and solvent evaporation the residues were reconstituted in 50μL of an 80:20 mixture of solvents A and B for the second-dimension HILIC analysis.
The second dimension HILIC analysis was performed on a 150 × 2.1mm HILIC Atlantis column with a particle size of 3μm (Waters, Etten-Leur, The Netherlands). Here solvent A consisted of 0.1% formic acid in acetonitrile and solvent B of 10mmol L−1 aqueous ammonium acetate solution+0.1% formic acid in Milli-Q water. The separation was performed at 40°C, the maximum temperature specified by the manufacturer, and a flow rate of 0.2mL min−1. The initial eluent composition was 95% A. A linear gradient was used to 70% B in 30min, followed by a linear gradient to 90% B in 5min where it was kept for another 5min. At the end of the run the eluent was reduced to 95% C in 2min. The eluent flow was then increased to 0.3mL min−1 in 1min. This flow rate was maintained for another 7min. Finally the flow was reduced to 0.2mL min−1 in 1min. and the system was allowed to equilibrate for 3min. The total run time was 54min. 45μL of each fraction collected from the ODS3 column was injected. At the outlet of the second dimension HILIC column fractions of 200μL were collected in 96-well plates. The fractions were neutralized, evaporated, and transferred to the activity assay (next section).
At-line Matsui assay
ACE inhibition by the collected fractions was measured in a 96-well plate assay according to the at-line method described in an earlier publication [1]. The method is based on the conversion of hippuryl–histidyl–leucine (HHL) into hippurate (H) and histidyl–leucine (HL) by the angiotensin I-converting enzyme (ACE). Briefly, after evaporation of the eluent the fractions were reconstituted in 50μL PBS buffer and vortex mixed for 1min. This solution (40μL) was used for the at-line assay. Percentage inhibition was calculated from the peak areas of HHL and H at 280nm. For the active fractions the remaining 10μL was used for identification of the peptides.
Structural identification of ACEI peptides
Peptide identification was performed on the QTOF MS instrument using HPLC–MS and MS–MS in the accurate mass mode. Fractions collected after the second dimension that showed activity were separated on the HILIC column now coupled to the MS using the gradient described above. The source and desolvation temperatures were 100°C and 300°C, respectively. The cone and desolvation gas flows were 100L h−1 and 800L h−1, respectively. The capillary voltage was 4kV and the cone voltage 35V. The collision energy was 10eV in full-scan mode and 25eV in MS–MS mode. The collision gas was argon and the analyser pressure was 4 × 10−5 mbar. In the lock spray probe a mixture of PEG 300, PEG 600, and PEG 1000 dissolved in 10mmol L−1 ammonium acetate in methanol–water 1:1 (v/v) at concentrations of 0.01, 0.02, and 0.04μg mL−1, respectively, was used. The reference flow was 5μL min−1. Spectra were recorded at a resolution of 10,000. Masslynx software (Waters) was used for data acquisition and evaluation.
Results and discussion
In a reversed-phase fractionation of a protein hydrolysate a significant percentage of the activity can be present in the unretained hydrophilic fraction. To be able to calculate the contribution of this fraction of our milk hydrolysate to the total activity, an ACE-inhibition profile of the entire product was produced. To this end 20μL of a 20mg mL−1 solution of the milk hydrolysate in solvent A was injected on to the 300mm ODS3 column. Fractions of 200μL each were collected from 0 to 65min, with an interval time of 1min, in a 96 well plate. The ACE inhibition of each fraction was determined. Figure 1a shows the MS TIC of the fermented milk and Fig. 1b shows the activity profile of the same analysis. From the data in Fig. 1b it was calculated that the hydrophilic compounds in fractions 6 to 15 represent approximately 25% of the total ACE inhibition. MS–MS showed that the high activity in fractions 27 to 31 resulted from the known ACE inhibiting peptides IPP and LPP. The activity in fractions 21 to 23 resulted from the peptide VPP. These peptides have been identified to be the main contributors to the ACE-inhibition of a Lactobacillus helveticus-fermented milk [2] and represent 40% of the total ACE inhibition. The remaining activity between fractions 16 and 40 is most likely caused by a mixture of ACE-inhibiting peptides of different chain lengths. This is still under investigation. In the region from fraction 6 to fraction 15 a large number of peptides co-elute and the spectra of the fractions are highly complex. Figure 2, for example, shows the mass spectrum of fraction 7 collected from the first dimension ODS 3 columns and the ion traces of three of the ACE-inhibiting peptides discussed later in this manuscript. At least 16 ions can be observed with intensities above 10% of the base peak, all representing different peptides. In order to be able to assign the measured activity to one or more of these peptides a further separation in an additional chromatographic dimension is required. To achieve this, fractions collected from the first-dimension ODS column were injected on the second dimension HILIC column. Again 200-μL fractions were collected in 96-well plates. After neutralization and evaporation the residues were reconstituted in 50μL Milli-Q water. Of this, 40μL was used for ACE inhibition testing, the remaining 10μL for MS identification. As an example of the results of these experiments, Fig. 3 shows the TIC chromatogram obtained from full-scanning analysis of fraction 7 of the ODS3 columns, now separated on the second dimension HILIC column. The TIC chromatogram clearly shows that the composition of this fraction is indeed very complex. To identify the peptide(s) responsible for the ACE-inhibition, the fractions collected in the 96-well plate were subjected to the ACE-inhibition assay. Figure 4 displays the activity distribution of ODS fraction 7. The data in Fig. 4 are corrected for the background recorded using a blank gradient, i.e. a gradient without injection. Blank analysis of the HILIC column showed slightly negative values compared with a reference of pure PBS. The reason for these negative responses is still unknown. The results in Fig. 4 clearly show the strong activity in HILIC fractions 17 to 19 and somewhat lower activity in fractions 12 to 16 from the HILIC column. LC–MS separations were performed on these fractions and the MS data were searched for peptides in the two active regions. In Fig. 5 the mass spectrum of fraction 18 is given. In the spectrum three abundant ions can be observed. A library search and MS–MS measurements revealed that m/z 272.1721 represents the molecular ion of the di-peptide RP (−0.7ppm), while m/z 253.1190 represents the molecular ion of HP (−2.1ppm) and m/z 235.1305 the loss of water from m/z 253.1190. Identification of the peptides in the remaining fractions of the HILIC column showed that fraction 7 of the first-dimension ODS3 column consisted of three free amino acids, 19 di-peptides and 2 tri-peptides while six compounds remained unidentified. The positively identified amino acids were E, Q, and K and the di-peptides were QD, KY, ER, RE, KP, HP, RP, AP, VK, EK, EW, and PH. The di-peptides ET, TP, TQ, PQ, KV, KE, and HK and the tri-peptides APK and VRG were tentatively identified. Structure confirmation was based on elution time, measured exact mass (error <5ppm) and MS–MS fragmentation pattern compared with those of the model compounds. Using this method the remaining hydrophilic fractions 5 to 15 collected from the ODS3 column were also analysed in the 2D mode. In Fig. 6 a three-dimensional plot of the activity distribution over the fractions of the two columns is given. In total, five amino acids, 35 di-peptides, 13 tri-peptides, one penta-peptide and 18 not yet identified compounds were found. The identities of all amino acids and the sequence of 27 di-peptides were again confirmed by use of model compounds. Table 1 lists the sequences of the identified peptides together with their reported IC50 values and ACE inhibition data at 20μmol L−1 established in house (Foltz et al. manuscript in preparation). For peptides for which no model compounds were available, identification was based solely on the fragmentation pattern in MS–MS.

LC separation of a 20 mg mL−1 solution of the milk hydrolysate powder on the ODS-3 reversed-phase column. a MS-TIC chromatogram. b Activity profile. Error bars indicate the standard deviation (n = 3). Fraction volume 200 μL

Mass spectrum of fraction 7 collected from the first-dimension ODS3 column

MS-TIC of fraction 7 from the ODS3 column analysed on the HILIC column. The inset shows the selected ion traces of three of the most active peptides

ACEI profile of fraction 7 from the ODS3 column analysed on the HILIC column

Mass spectrum of fraction 18 collected from the HILIC column

Three-dimensional display of the ACEI distribution of the fractions collected from the ODS3 column and the HILIC column
To verify whether the identified peptides indeed fully explain the measured activity of the hydrophilic fraction the concentration of all peptides should be determined. Next a mixture should be prepared which is then analysed using the two-dimensional method described in this article. The ACE-inhibition profile of this mixture should be identical with that of the sample. Such an experiment will hardly be feasible because it requires the availability of all the components. To estimate of the completeness of identification, the contribution of each peptide was estimated from the measured peak area and the IC50 value obtained from literature or from unpublished in-house data. To estimate the contribution of an individual peptide the following relationship was used: CA ≈ A p/IC50p. In this equation CA represents the contribution of the peptide to the measured ACE-inhibition, A p the peak area, and IC50p the IC50 value of the peptide. Clearly this proportionality equation is only approximate as it assumes that the MS response for all peptides is identical. When both the peak area and the IC50 value are taken into consideration it can be concluded that the di-peptides AP, RP, VK, and EK are the main peptides responsible for the measured ACE-inhibition of the hydrophilic fractions 6 to 10 collected from the ODS3 column (Fig. 7). In Fig. 8 the activities of all peptides in each fraction are summarized and the profile is compared with that of the measured profile. The two profiles are similar. The differences between the graphs can be explained from the fact that no correction was applied for differences in MS responses of the various peptides.

Distribution of the ACE-inhibiting activity, calculated from Table 1, in hydrophilic fractions 5 to 10 collected from the ODS3 column

Calculated (bottom) and measured (top) ACE inhibition profiles
The remaining fractions 11 to 15 of the ODS3 column were analysed in the same way as fractions 6 to 10. These analyses showed that the peptide EW is responsible for the major part of the ACE-inhibition measured in these fractions. In total, the five peptides identified in the fractions 6 to 15 are responsible for approximately 85% of the activity measured in the hydrophilic fractions. The most important contribution is that of the di-peptide RP, which is responsible for 34%. Together with the peptides VPP, IPP, and LPP now approximately 65% of the total activity of the product has been explained. The remaining 35% is distributed over a large number of peptides with relatively low overall activities.
When compiling the list of published IC50 values for the peptides identified in this study in some cases large differences were seen for the IC50 values of proline-containing peptides in different publications. The reported IC50 values for AP, for example, vary between 29 and 269 μmol L−1 [14]. This large difference might be caused by variations in the cis-to-trans ratio of the proline present in synthetic model compounds as a result of differences in the synthetic routes (Fmoc or Boc), as was shown for the peptide DKIHP by Gómez-Ruiz [15]. In our calculations the trans-Pro value of 29 μmol L−1 for AP was used, because trans-Pro is known to be dominant in natural products [15]. Many of the identified di-peptides were found in milk hydrolysates for the first time. For most of the newly identified ACE active peptides it is actually the first time they are reported at all. The long list of newly identified peptides found here clearly demonstrates the potential of the two dimensional separation approach of HILIC and reversed-phase HPLC described here.
Conclusions
Two-dimensional liquid chromatography in combination with mass spectrometry was successfully used for identification of poorly retained peptides present in enzymatically hydrolysed milk protein. A standard C18 reversed-phase column was used for the first separation followed by a second dimensional separation on a HILIC column. This two-dimensional procedure significantly improves the separation of hydrophilic peptides that elute almost unretained on a reversed-phase column and co-elute with numerous other compounds such as carbohydrates and salts. These compounds suppress the MS ionization and complicate identification. The method enabled the identification of hydrophilic peptides in complex mixtures. In the hydrophilic fraction of the milk hydrolysate investigated 71 compounds were found, including five free amino acids, 35 di-peptides, 12 tri-peptides, one penta-peptide, and 18 not yet identified compounds. Five peptides, RP, AP, VK, EK, and EW were responsible for approximately 85% of the measured activity of the hydrophilic fraction; of these RP made the highest contribution of 34%. None of the five peptides has, to our knowledge, been reported earlier in milk hydrolysates or related products such as yogurt or cheese. Most of the peptides involved remained undetected in single-dimensional chromatography.
References
Nakamura Y, Yamamoto N, Sakai K, Okubo A, Yamazaki S, Takano T (1995) J Dairy Sci 78:777–783
Lopez-Exposito I, Quiros A, Amigo L et al (2007) Lait 87:241–249
Tsai JS, Chen TJ, Pan BS et al (2008) Food Chem 106:552–558
van Platerink CJ, Janssen HGM, Haverkamp J (2007) J Chromatogr B 846:147–154
Robert M-C, Razaname A, Mutter M, Juillerat MA (2004) J Agric Food Chem 52:6923–6931
Maeno M, Yamamoto N, Takano T (1996) J Dairy Sci 79:1316–1321
Gómez-Ruiz JA, Taborda G, Amigo L, Reccio I, Ramos M (2006) Eur Food Res Technol 223:595–601
Alpert AJ (1990) J Chromatogr 499:177–196
Daunoravicius Z, Juknaite I, Naujalis E (2006) Chromatographia 63:373–377
Jandera P, Fischer J, Lahovska H, Novotna K, Cesla P, Kolarova L (2006) J Chromatogr A 1119:3–10
Wang XD, Li WY, Rasmussen HT (2005) J Chromatogr A 1083:58–62
Yoshida T (2004) J Biochem Biophys Methods 60:265–280
Schlichtherie-Cerny H, Affolter M, Cerny C (2003) Anal Chem 75:2349–2354
Wu J, Aluko RE, Nakai S (2006) J Agric Food Chem 54:732–738
Gómez-Ruiz JA, Recio I, Belloque J (2004) J Agric Food Chem 52:6315–6319
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van Platerink, C.J., Janssen, HG.M. & Haverkamp, J. Application of at-line two-dimensional liquid chromatography–mass spectrometry for identification of small hydrophilic angiotensin I-inhibiting peptides in milk hydrolysates. Anal Bioanal Chem 391, 299–307 (2008). https://doi.org/10.1007/s00216-008-1990-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-1990-3