
Abstract
In nonreversible hydroformylations, the computational evaluation of regio- and stereoselectivities from the relative energy barriers of the transition states (TS) for the alkyl-Rh intermediate formation step is possible, provided all low energy conformers are considered. In contrast, in reversible hydroformylations, also the subsequent reaction steps need to be taken into account to shed some light on mechanistic details. Thus, an extensive comparison of branched (B) and linear (L) reaction pathways for the Rh-catalyzed hydroformylation of 3,4,4-trimethylpent-1-ene (a bulky chiral substrate), going from a number of reactant complexes to products, has been carried out to rationalize the experimental result that pointed to reaction reversibility, although the value of the regioselectivity ratio (B:L = 15:85), based on alkyl-Rh TS free energies, computed under the hypothesis of nonreversibility, was in satisfactory agreement with the experimental one (5:95). A density functional theory approach at the B3P86/6-31G* level coupled to effective core potentials for Rh in the LanL2DZ valence basis set has been employed. By comparing the activation free energies involved in the various steps for the different reactant adducts, interestingly a similar behavior along all the linear pathways is found: the alkyl-Rh formation TS presents the highest barrier; thus, the reaction is nonreversible for all the linear isomers that invariably proceed to yield the linear aldehyde. Conversely, the behavior is quite different along the branched pathways. While some branched isomers eventually produce the corresponding aldehydes, two of the others follow distinct competing pathways, because β-hydride elimination occurs (a) to the terminal olefin-Rh complex (the starting material) that reacts again with the original regioselectivity, increasing the linear fraction, when the CO addition and insertion TS are higher than the alkyl-Rh TS and (b) to the internal olefin-Rh complex when the CO addition and insertion TS are higher than the relevant β-hydride elimination TS, but not than the alkyl-Rh TS. The solvent effect on the reversible profile, evaluated either in the supermolecule approach by adding a benzene molecule to the calculations or in the IEF-PCM framework (ε = 2.247), does not bring about any substantial change in the profile, leaving unaltered the conclusions reached.


























Similar content being viewed by others
Notes
Notice that to simplify the notation, only B3P86/6-31G* is reported throughout.
Following the suggestion by Jonas and Thiel [46], who systematically investigated the performance of a variety of basis sets and methods (HF, DFT, and MP2) in transition metal carbonyls but Rh ones, we initially adopted BP86. Tests carried out in Ref. [28] on hydroformylations with Rh carbonyls supported the use of B3 instead.
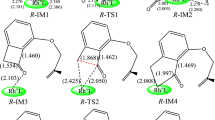
Occasionally, the CO groups in the plane roughly perpendicular to H–Rh–CO can swap their positions, thus inverting the pyramid orientation.
Interestingly enough, the potential- and free-energy-based diastereoselectivity ratios (b:b′) are conversely very similar to each other, that is, 78:22 (ΔE) and 79:21 (ΔG); the difference between potential- and free-energy-based regioselectivity ratios thus depends on the linear regioisomers. Using a formula analogous to Eq. 2 and replacing b and b′ with the computationally available l and l′ populations, the two l:l′ potential- and free-energy-based diastereoselectivity ratios actually turn out to be 59:41 and 86.3:13.7, respectively.
Thus two species react to give just one new compound.
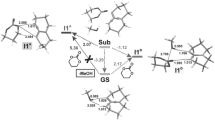
For branched structures, out of the two with a suitable orientation.
The intermediate corresponding to “TSH2 b′2 in p” is “H2Int-b′2 in p” (reported in Table 6 and displayed in Fig. S8) that evolves to the branched aldehyde through “TS–H2 b’2 in p” (∆G=15.13 kcal/mol), that is, with a much higher barrier than when the hydrogens are in the basal CO plane.
The most critical case because of TS relative values, involving β-elimination leading either to internal or initial olefin.
References
Roelen O (1938) (Ruhrchemie AG). D. B. P. 849 458
Roelen O (1953) Chem Zentr 927–929
Cornils B, Herrmann WA (1996) Applied homogeneous catalysis with organometallic compounds, vol 1. Wiley, New York, pp 3–25
Papadogianakis G, Sheldon RA (1996) New J Chem 20:175–185
Süss-Fink G, Meister G (1993) Adv Organomet Chem 35:41–134
Versluis L, Ziegler T, Fan L (1990) Inorg Chem 29:4530–4536
Wender I, Pino P (1977) Organic syntheses via metal carbonyls, vol 2. Wiley, New York
Falbe J (1980) New nyntheses with carbon monoxide. Springer, Berlin
Pignolet LH (ed) (1983) Homogeneous catalysis with metal phosphine complexes. Plenum, New York
Casey CP, Petrovich LM (1995) J Am Chem Soc 117:6007–6014
Casey CP, Paulsen EL, Bettenmueller EW, Proft BR, Matter BA, Powell DR (1999) J Am Chem Soc 119:11817–11825
Van Leeuwen PWNM, Kamer PCJ, Reek JNH (1999) Pure Appl Chem 71:1443–1452
Nozaki K, Sakai N, Nanno T, Higashijima T, Mano S, Horiuchi T, Takaya H (1997) J Am Chem Soc 119:4413–4423 and refs quoted therein
Herrmann WA, Kohlpaintner CW, Herdtwck E, Kiprof P (1991) Inorg Chem 30:4271–4275
Herrmann WA, Schmid R, Kohlpaintner CW, Priermeier T (1995) Organometallics 14:1961–1968
Decker SA, Cundari TR (2001) Organometallics 20:2827–2841
Decker SA, Cundari TR (2001) J Organomet Chem 635:132–141
Decker SA, Cundari TR (2002) New J Chem 26:129–135
Rocha WR, De Almeida WB (2000) Int J Quantum Chem 78:42–51
Dias RP, Prates MSL Jr, De Almeida WB, Rocha WR (2011) Int J Quantum Chem 111:1280–1292
Carbó JJ, Maseras F, Bo C, van Leeuwen PW (2001) J Am Chem Soc 123:7630–7637
Landis CR, Uddin J (2002) J Chem Soc, Dalton Trans 729–742
Beller M, Cornils B, Frohning CD, Kohlpaintner CW (1995) J Mol Cat A Chem 104:17–85
Pidun U, Frenking G (1998) Chem Eur J 4:522–540
Torrent M, Solà M, Frenking G (2000) Chem Rev 100:439–493 and refs quoted therein
Consiglio G, Pino P (1982) Top Curr Chem 105:77–123
Gleich D, Schmid R, Herrmann WA (1998) Organometallics 17:4828–4834
Alagona G, Ghio C, Lazzaroni R, Settambolo R (2001) Organometallics 20:5394–5404
Alagona G, Ghio C, Lazzaroni R, Settambolo R (2004) Inorg Chim Acta 357:2980–2988
Alagona G, Ghio C (2005) J Organomet Chem 690:2339–2350
Settambolo R, Rocchiccioli S, Lazzaroni R, Alagona G (2006) Lett Org Chem 3:10–12
Alagona G, Ghio C, Rocchiccioli S (2007) J Mol Model 13:823–837
Lazzaroni R, Settambolo R, Marchetti M, Paganelli S, Alagona G, Ghio C (2008) Inorg Chim Acta 362:1641–1644
Ghio C, Lazzaroni R, Alagona G, (2009) Eur J Inorg Chem 98–103
Lazzaroni R, Settambolo R, Alagona G, Ghio C (2010) Coord Chem Rev 254:696–706. Erratum in: (2011) Coord Chem Rev 255:3031
Alagona G, Lazzaroni R, Ghio C (2011) J Mol Model 17:2275–2284. Erratum. doi:10.1007/s00894-012-1373-8
Lazzaroni R, Settambolo R, Alagona G, Ghio C (2012) J Mol Cat A Chem. doi:10.1016/j.molcata.2011.12.021
McQuarrie DA (2000) Statistical mechanics. University Science Book, Sausalito
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision C.02, Gaussian Inc., Wallingford
Becke AD (1993) J Chem Phys 98:5648–5652
Perdew JP (1986) Phys Rev B 33:8822–8824
Hehre WJ, Radom L, Schleyer PVR, Pople JA (1986) Ab initio molecular orbital theory. Wiley, New York
Dunning TH Jr, Hay PJ (1976) In: Schaefer HF III (ed) Modern theoretical chemistry. Plenum, New York, pp 1–28
Hay PJ, Wadt WR (1985) J Chem Phys 82:270–283
Hay PJ, Wadt WR (1985) J Chem Phys 82:299–310
Jonas V, Thiel W (1995) J Chem Phys 102:8474–8484
Goh SR, Marynick DS (2002) Organometallics 21:2262–2267
Frenking G, Antes I, Boehme M, Dapprich S, Ehlers AW, Jonas V, Nehaus A, Otto M, Stegmann R, Veldjamp A, Vyboishchikov SF (1996) In: Lipkowitz KB, Boyd DB (eds) Reviews in computational chemistry, vol 8. VCH, New York, pp 63–144
Tomasi J, Persico M (1994) Chem Rev 94:2027–2094
Barone V, Cossi M, Tomasi J (1997) J Chem Phys 107:3210–3221
Cancès E, Mennucci B, Tomasi J (1997) J Chem Phys 107:3032–3041
Cancès E, Mennucci B (1998) J Chem Phys 109:249–259
Cancès E, Mennucci B, Tomasi J (1998) J Chem Phys 109:260–266
Amovilli C, Barone V, Cammi R, Cancès E, Cossi M, Mennucci B, Pomelli CS, Tomasi J (1998) Adv Quantum Chem 32:227–261
Tomasi J, Mennucci B, Cammi R (2005) Chem Rev 105:2999–3094
Tomasi J (2007) In: Mennucci B, Cammi R (eds) Continuum solvation models in chemical physics: from theory to applications. Wiley, New York, pp 1–28
Schaftenaar G, Noordik JH (2000) J Comput Aided Mol Design 14:123–134
Curtin DY (1954) Rec Chem Prog 15:111–128
Hammett LP (1970) Physical organic chemistry, 2nd edn. McGraw-Hill, New York, pp 119–120 Chapter 5
Seeman JI (1983) Chem Rev 83:84–134
Matsubara T, Koga N, Ding Y, Musaev DG, Morokuma K (1997) Organometallics 16:1065–1078
Yagupsky G, Brown CK, Wilkinson G (1969) J Chem Soc Chem Comm 1244–1245
Yagupsky G, Brown CK, Wilkinson G (1970) J Chem Soc A 1392–1401
Tang D, Qin S, Su Z, Hu C (2007) Organometallics 26:33–47
Nowroozi-Isfahani T, Musaev DG, McDonald FE, Morokuma K (2005) Organometallics 24:2921–2929
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to Professor Vincenzo Barone and published as part of the special collection of articles celebrating his 60th birthday.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Alagona, G., Ghio, C. The fate of branched and linear isomers in the rhodium-catalyzed hydroformylation of 3,4,4-trimethylpent-1-ene. Theor Chem Acc 131, 1142 (2012). https://doi.org/10.1007/s00214-012-1142-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-012-1142-x