
Abstract
Omalizumab is an effective therapeutic humanized murine IgE antibody in many cases of primary systemic mast cell activation disease (MCAD). The present study should enable the clinician to recognize when treatment of MCAD with omalizumab is contraindicated because of the potential risk of severe serum sickness and to report our successful therapeutic strategy for such adverse event (AE). Our clinical observations, a review of the literature including the event reports in the FDA AE Reporting System, the European Medicines Agency Eudra-Vigilance databases (preferred search terms: omalizumab, Xolair®, and serum sickness) and information from the manufacturer’s Novartis database were used. Omalizumab therapy may be more likely to cause serum sickness than previously thought. In patients with regular adrenal function, serum sickness can occur after 3 to 10 days which resolves after the antigen and circulating immune complexes are cleared. If the symptoms do not resolve within a week, injection of 20 to 40 mg of prednisolone on two consecutive days could be given. However, in MCAD patients whose adrenal cortical function is completely suppressed by exogenous glucocorticoid therapy, there is a high risk that serum sickness will be masked by the MCAD and evolve in a severe form with pronounced damage of organs and tissues, potentially leading to death. Therefore, before the application of the first omalizumab dose, it is important to ensure that the function of the adrenal cortex is not significantly limited so that any occurring type III allergy can be self-limiting.
Similar content being viewed by others


Introduction
Omalizumab has become increasingly important in the treatment of diseases (e.g., allergic asthma, chronic urticaria, mast cell activation disease) where there is increased activation of mast cells (Foroughi et al. 2007; Thomson and Chaudhuri 2012; Incorvaia et al. 2014; Stokes 2017; Peterson and Coop 2017; Kavati et al. 2019). This medication has US Food and Drug Administration (FDA) and European Union (EU) approval for treatment of IgE-induced asthma and in chronic idiopathic urticaria. Particularly, in the case of primary systemic mast cell activation disease (MCAD), which in many cases is challenging to treat, omalizumab has proven useful in decreasing symptom intensity (Molderings et al. 2011, further references therein; Zampetti 2018; Broesby-Olsen et al. 2018; Slapnicar et al. 2019; Lemal et al. 2019). Nonetheless, we show here that in certain circumstances, omalizumab may pose a risk for serum sickness. Our clinical observations, a review of the literature including the event reports in the FDA Adverse Event Reporting System, the European Medicines Agency Eudra-Vigilance databases (preferred search terms: omalizumab, Xolair®, and serum sickness) and information from the manufacturer’s Novartis database were used in the present analysis.
The aims of this study are to enable the clinician to recognize when a treatment of mast cell–related disease with omalizumab is contraindicated because of the potential risk of severe serum sickness (i.e., steroid use with resulting adrenal insufficiency) and to report our successful therapy strategy for such adverse event, since no evidence-based guidelines exist for the treatment of severe serum sickness.
Mast cells, systemic mast cell activation disease (MCAD), and its therapy
Mast cells are hematopoietic tissue immune cells that act both as effector and regulatory cells (Afrin et al. 2016). They play central roles in adaptive and innate immunity (e.g., Cardamone et al. 2016). This versatility is reflected in the myriad of immunologic and non-immune activation stimuli (e.g., by G protein-coupled receptors) resulting in the secretion of a large number (> 1000) of pre-stored mediators (e.g., histamine, tryptase, and numerous de novo–synthesized lipid mediators), chemokines, and cytokines (Ibelgaufts 2019). The profile of such mediators can markedly differ between and within organs/tissues, depending upon the micro-environmental factors and/or the nature of the stimulus (e.g., Marshall et al. 2003).
MCAD (prevalence up to 17% [Molderings et al. 2013; Lyons et al. 2016; Lazaridis and Germanidis 2018]) comprises a heterogeneous group of multifactorial disorders characterized by epigenetic and genetic alterations (somatic and germline mutations) in a variety of genes inducing an inappropriate release of variable subsets of mast cell mediators together with accumulation of either morphologically altered and immunohistochemically identifiable mutated mast cells (systemic mastocytosis and mast cell leukemia) or alternatively, morphologically ordinary mast cells due to impaired apoptosis (mast cell activation syndrome and well-differentiated systemic mastocytosis) (Afrin et al. 2016; Online Resource 1).
MCAD can affect single and multiple systems (i.e., organs and tissues; Theoharides et al. 2015), usually manifesting with symptoms in a subacute or chronic waxing/waning or recurrent manner (Afrin et al. 2017, further references therein). Due to both the widespread distribution of mast cells and the great heterogeneity of aberrant mediator expression patterns, symptoms can involve virtually all organs and tissues; hence, the clinical presentation of MCAD is very diverse (Online Resource 2).
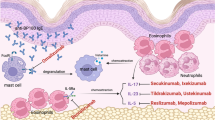
Currently, there is no cure for primary mast cell disorders. As the dominant feature of MCAD is inappropriate increased and unregulated mast cell activation, treatment invariably involves trigger identification and avoidance, respectively, plus control of mast cell mediator production and action. Generally, less expensive interventions (e.g., histamine H1 and H2 receptor antagonists, leukotriene receptor antagonists) are tried first, but often symptoms persist. Overall, the medical treatment strategy is a stepwise approach to manage the increased mast cell activity (Fig. 1; for a comprehensive review of the current and potential future treatment options of MCAD, see Molderings et al. 2016).

Therapeutic procedure in mast cell activation disease (modified from Molderings et al. 2016)
Omalizumab
Omalizumab is a recombinant 95% humanized IgG1κ monoclonal antibody (mAb; 5% of murine origin mainly in the Fab fragment; Presta et al. 1993, 1994) which recognizes and binds with picomolar affinity to the third constant domain of the IgE heavy chain (Cε3) of circulating IgE (Vichyanond 2011; Pennington et al. 2016; Davies et al. 2017; further references therein). Cε3 (even more so the Cε3–4 portion) is the docking site that normally binds electrostatically to the α2 subunit of the high-affinity (FcεRI) and low-affinity (FcεRII) IgE receptor on mast cells, basophils, and other cell types. Omalizumab forms immune complexes (ICs) with free IgE and, thus, prevents its interaction with both receptors. An important property of omalizumab is that it neither interacts with cell-bound IgE, nor activates mast cells or basophils (Belliveau 2005). Following subcutaneous administration, omalizumab is absorbed slowly (linearly above doses > 0.5 mg/kg), reaching peak serum concentrations after an average of 7–8 days [Omalizumab, Xolair, label information at www.fda.gov/cder/foi/label/2003/omalgen062003LB.pdf.]. The omalizumab-IgE complexes are cleared via interactions with Fcγ receptors of the hepatic sinusoidal and other endothelial cells of the reticuloendothelial system (Ghetie et al. 1996; Mariani and Strober 1990). Noteworthy, the clearance of the free mAb itself is slow (mean 2.4 ± 1.1 ml kg−1 day−1), with a terminal half-life (t1/2) of 26 days (Omalizumab, Xolair, label information at www.fda.gov/cder/foi/label/2003/omalgen062003LB.pdf; Lowe et al. 2009). According to general pharmacokinetic principles, elimination is near-complete after five half-lives of a drug. However, it has been observed that omalizumab may put a disease into clinical remission for up to 6 years after drug discontinuation (Nopp et al. 2007, 2010; Molimard et al. 2014; Bölke et al. 2019), which may imply that the specific ICs remain in circulation long after discontinuation of treatment (Starke P 2016 Clinical Review. BLA 103576 S-5225 - Xolair [Omalizumab]; further references therein). As omalizumab targets a specific component of the immune system, therapy may have the potential to increase the risk of immune disorders. The mAb component of murine origin may induce allergic reactions (for further serious adverse effects, see Online Resource 3).
Type III hypersensitivity reactions
Type III hypersensitivity reactions (Online Resource 4), also termed Immune Complex reactions, are mediated by IgM and IgG antibodies which react with soluble antigens (including allogenic/chimeric antibodies, such as omalizumab) forming ICs of different sizes (Shmagel and Chereshnev 2009). Serum sickness and serum sickness–like reactions are delayed type III hypersensitivity clinical expressions (Online Resource 4) becoming evident usually 3 to 10 days after exposure to the antigen, when antibodies have been sufficiently generated against the foreign protein and have formed ICs with these circulating antigens. Multimeric antigen-antibody complexes are efficient activators of the complement cascade through its classical pathway. Damage results from the action of cleaved complement anaphylatoxins C3a and C5a (that can be detected by decreased levels of circulating C3 and C4), which, respectively, mediate the induction of granule release from mast cells and recruitment of inflammatory cells leading to tissue damage through phagocytosis by neutrophils and macrophages. Tissue injury typically involve kidney, skin, and mucous membranes (Online Resource 4). The frequency (Online Resource 5) and severity of serum sickness depends on the composition of the antibody, the size of the ICs, and the functionality of the adrenal gland. As long as the adrenal gland is able to produce the amount of cortisol necessary to suppress the inflammatory immune response, serum sickness remains a self-limited disease which will usually resolve upon discontinuation of the offending agent.
The diagnosis of serum sickness is primarily based on patient history, including recent medications but also can be suspected by specific symptoms (Online Resource 4) and laboratory data, including leukocytosis, mild thrombocytopenia, elevated acute-phase proteins such as C-reactive protein (CRP) and Factor VIII, low C3 and C4 complement factors, decreased total hemolytic complement (CH50), and elevated circulating IC, as detected by C1q consumption. Laboratory results may be widely variable and contribute to the diagnosis only if they are positive.
Use of omalizumab in the treatment of systemic mast cell disease
Omalizumab is an effective therapeutic drug in many cases of MCAD (for references, see Introduction). The initial hope that the risk of AEs would be lower for omalizumab than for other chimeric antibodies, because of humanization procedures and the selective binding with IgE, has not been met (Online Resource 5). In particular, the type III allergy serum sickness has been assumed to be a rare AE of omalizumab therapy. Although its real prevalence can only be roughly estimated, the fact that this AE has been repeatedly reported in various studies with small to medium numbers of patients argues against it being a rare event (Table 1). This assumption is supported by the number of reports in the spontaneous reporting system of the FDA and EMA (Table 1). For other drugs entered in the same databases, it is estimated that only 1% to 10% of all AEs have ever been reported (Rogers et al. 1988; Scott et al. 1987). Whether the same is true of omalizumab-induced serum sickness remains unclear.
Thus, it is important to consider several factors before deciding to use omalizumab in MCAD patients. In particular:
Patients with normal adrenal function
At present, omalizumab is a third-line treatment option for MCAD (Fig. 1), which in some patients improves symptoms, reduces the number of flares, and strikingly alleviates pain intensity, in particular, in those MCAD patients with pain as a dominant symptom (Molderings et al. 2011; further references therein). Although omalizumab has been reported as a well-tolerated agent (Stokes 2017; Broesby-Olsen et al. 2018), transient mild to moderate mast cell mediator–induced symptoms occurring within several hours after injection have been observed, suggesting possible activation of mast cells, possibly outweighing the desired pharmacological effect of omalizumab (Molderings et al. 2011). This implies that when the triggering of mast cell mediator release (the mechanism of triggering has still to be identified, since the drug is thought to bind only to free IgE) occurs after any of the first three injections, or even becomes more intense from injection to injection, the treatment with omalizumab should be stopped immediately. In the worst case, serum sickness with IC formation, cytokine release, and intense mast cell mediator–induced symptoms may occur after 3 to 10 days; this reaction resolves after the antigen and ICs are cleared. If the symptoms do not resolve within a week, injection of 20 to 40 mg of prednisolone on two consecutive days could be given to stop serum sickness.
Patients with complete adrenal insufficiency
Usually, serum sickness would not have such a threatening nature unless three conditions typically found in MCAD can mask and/or turn it into a serious AE:
-
(1)
The symptoms of MCAD (Online Resource 2) can be indistinguishable from those of serum sickness. Therefore, an omalizumab-induced serum disease may not be recognized even for a long time in these patients, i.e., until the occurrence of complement-related organ and tissue failures which are less common in MCAD suggests the concomitant presence of serum sickness.
-
(2)
The regular administration of 150 mg or 300 mg omalizumab in 2- to 4-week intervals is more prone to change a usually self-limiting type III allergy into a more serious AE, due to a progressive and significant activation of the complement system showing more persistent and destructive properties.
-
(3)
MCAD patients, especially those with first- and second-line-resistant therapy, aggressive or advanced MCAD, are also treated with glucocorticosteroids at doses (as a rule with prednisone > 20 mg/day) above the Cushing dose (commonly defined as prednisone equivalent > 7.5 mg/day) (Akin 2014; Quintas-Cardama et al. 2006; Valent et al. 2005; Zen et al. 2011; Valent et al. 2012; Afrin et al. 2016). The chronic administration of glucocorticoids in such doses results in a complete loss of endogenous cortisol production by the cells in the zona fasciculata of the adrenal cortex. If serum sickness develops in a MCAD patient treated with such glucocorticoid doses and omalizumab, the serum sickness symptoms would be additionally masked/delayed by the immunosuppressive effect of the exogenous glucocorticoid, without decisively preventing at this dosage the activation of the complement system. When in such a patient the exogenous glucocorticoid dose is tapered off according to the applicable time frame (e.g., Pavlicek 2014), e.g., because the therapy of MCAD is switched to another immunosuppressive agent (e.g., kinase inhibitor), pharmacokinetics indicates a high probability that a flare of serum sickness will occur due to massive complement activation with possible consequent organ failures [as seen with occurrence of Churg-Strauss syndrome (Giavina-Bianchi et al. 2007; Pabst et al. 2008; Ruppert et al. 2008; Jachiet et al. 2016)]. In the absence of a prompt intensive medical treatment and/or misunderstanding of the cause, this flare can be fatal (Table 1). In this situation, besides ensuring support of vital signs, it is crucial to suppress the activation of the complement system with high doses of glucocorticosteroids. There are no evidence-based guidelines for the daily glucocorticoid dose to be used; hence, different daily doses of up to 1 g prednisone are reported in the literature (Pilette et al. 2007; Kumar 2019). We have decided to start probatorily in our patients with prednisone 80 mg/day which turned out as enough for successful treatment, followed by symptom-adapted reductions in prednisone dose, which should take into consideration the peculiarity of omalizumab pharmacokinetics. As said, the average biological half-life of this mAb is about 26 days, which means that only after 5 months (i.e., five half-life periods) would the original compound be eliminated from the body. More importantly, omalizumab ICs may circulate in the body for more than 1 year after the substance has been discontinued (Chang 2000; Tridente 2014; Starke 2015). It is not known whether these ICs are able to sustain serum sickness or other AEs. Since a detection assay of omalizumab-specific circulating ICs is not available commercially, the duration of therapy can only be based on the degree of the symptoms of complement activation when tapering the initial dose of prednisone. However, the differential identification of symptoms is difficult because of their extensive overlap with MCAD symptoms. Of note, the parameters for detecting complement activation (see above) may be initially pathologically altered, but the changes are short-lasting. As for the patients we have treated (Table 1, first row), the time to resolution of severe serum sickness was up to 15 months. At least one patient had significant irreversible disorders. Whether the less severe residual disorders will be irreversible in the second surviving patient remains to be seen.
Conclusions
Omalizumab therapy may be more prone to cause serum sickness than previously thought. Indeed, in a patient whose adrenal cortical function is completely suppressed by exogenous glucocorticoid therapy for the treatment of the underlying disease (such as MCAD, long-term inhaled corticosteroid therapy of asthma, chronic urticaria, and others), there is a higher risk that serum sickness will be masked and evolve into a severe, potentially fatal form with pronounced damage of organs and tissues. Since the diagnosis of serum sickness is essentially clinical (because the sensitivity of the laboratory parameters is unreliable, and the overlap of serum sickness symptoms with those of the omalizumab-treated underlying disease), recognition of serum sickness disease can be very difficult and often only occurs by chance. If there is a clinical suspicion of correlation of the occurrence of symptoms with omalizumab administration, it may be diagnostically useful to compare the values for the acute phase proteins CRP and Factor VIII determined shortly before the re-exposure with omalizumab and during the symptomatic period after re-exposure. Most importantly, before the application of the first omalizumab dose (and probably other antibodies too), it is necessary to ensure that the function of the adrenal cortex is not significantly limited (which can be excluded by determination of basal and adrenocorticotropic hormone-stimulated cortisol), so that any occurring type III allergy/serum sickness can be self-limiting.
References
Eapen A, Kloepfer KM (2018) Serum sickness-like reaction in a pediatric patient using omalizumab for chronic spontaneous urticaria. Pediatric Allergy Immunology 29:449–450
Afrin LB, Butterfield JH, Raithel M, Molderings GJ (2016) Often seen, rarely recognized: mast cell activation disease - a guide to diagnosis and therapeutic options. Ann Med 48:190–201
Afrin LB, Self S, Menk J, Lazarchick J (2017) Characterization of mast cell activation syndrome. Am J Med Sci 353:207–215
Akin C (2014) Mast cell activation disorders. J Allergy Clin Immunol: In Practice 2:252–257
Althin M (2018) Evaluation of Xolair® (omalizumab) therapy in patients treated at Örebro University Hospital 2006-2017. Örebro University Hospital, Örebro, Sweden, Medical Thesis
Belliveau PP (2005) Omalizumab: a monoclonal anti-IgE antibody. Med Gen Med 7:27
Berger W, Gupta N, McAlary M, Fowler-Taylor A (2003) Evaluation of long-term safety of the anti-IgE antibody, omalizumab, in children with allergic asthma. Annals of Allergy, Asthma & Immunology 91:182–188
Bölke G, Church MK, Bergmann KC (2019) Comparison of extended intervals and dose reduction of omalizumab for asthma control. Allergo J Int 28:1–4
Broesby-Olsen S, Vestergaard H, Mortz CG, Jensen B, Havelund T, Hermann AP, Siebenhaar F, Møller MB, Kristensen TK, Bindslev-Jensen C, Mastocytosis Centre Odense University Hospital (MastOUH) (2018) Omalizumab prevents anaphylaxis and improves symptoms in systemic mastocytosis: efficacy and safety observations. Allergy 73:230–238
Cardamone C, Parente R, Feo GD, Triggiani M (2016) Mast cells as effector cells of innate immunity and regulators of adaptive immunity. Immunol Lett 178:10–14
Chang TW (2000) The pharmacological basis of anti-IgE therapy. Nat Biotechnol 18:157–162
Chipps BE, Zeiger RS, Luskin AT, Busse WW, Trzaskoma BL, Antonova EN, Pazwash H, Limb SL, Solari PG, Griffin NM, Casale TB (2017) Baseline asthma burden, comorbidities, and biomarkers in omalizumab-treated patients in PROSPERO. Ann Allergy Asthma Immunol 119:524–532
Corren J, Casale TB, Lanier B, Buhl R, Holgate S, Jimenez P (2009) Safety and tolerability of omalizumab. Clin Exp Allergy 39:788–797
Davies AM, Allan EG, Keeble AH, Delgado J, Cossins BP, Mitropoulou AN, Pang MOY, Ceska T, Beavil AJ, Craggs G, Westwood M, Henry AJ, McDonnell JM, Sutton BJ (2017) Allosteric mechanism of action of the therapeutic anti-IgE antibody omalizumab. J Biol Chem 292:9975–9987
Dreyfus DH, Randolph CC (2006) Characterization of an anaphylactoid reaction to omalizumab. Ann Allergy Asthma Immunol 96:624–627
Foroughi S, Foster B, Kim N, Bernardino LB, Scott LM, Hamilton RG, Metcalfe DD, Mannon PJ, Prussin C (2007) Anti-IgE treatment of eosinophil-associated gastrointestinal disorders. J Allergy Clin Immunol 120:594–601
Ghetie V, Hubbard JG, Kim JK, Tsen MF, Lee Y, Ward ES (1996) Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. Eur J Immunol 26:690–696
Giavina-Bianchi P, Giavina-Bianchi M, Agondi R, Kalil J (2007) Administration of anti-IgE to a Churg-Strauss syndrome patient. Int Arch Allergy Immunol 144:155–158
Harrison RG, MacRae M, Karsh J, Santucci S, Yang WH (2015) Anaphylaxis and serum sickness in patients receiving omalizumab: reviewing the data in light of clinical experience. Annals of Allergy, Asthma & Immunology 115:77–78
Ibelgaufts H (2019) Mast cells. Cytokines Online Pathfinder Encyclopedia. http://www.cells-talk.com/index.php/page/copelibrary?key=mast%20cells, accessed October 9, 2019
Incorvaia C, Mauro M, Russello M, Formigoni C, Riario-Sforza GG, Ridolo E (2014) Omalizumab, an anti-immunoglobulin E antibody: state of the art. Drug Des Devel Ther 8:197–207
Jachiet M, Samson M, Cottin V, Kahn JE, Le Guenno G, Bonniaud P, Devilliers H, Bouillet L, Gondouin A, Makhlouf F, Meaux-Ruault N, Gil H, Bienvenu B, Coste A, Groh M, Giraud V, Dominique S, Godeau B, Puéchal X, Khouatra C, Ruivard M, Le Jeunne C, Mouthon L, Guillevin L, Terrier B, French Vasculitis Study Group (2016) Anti-IgE monoclonal antibody (omalizumab) in refractory and relapsing eosinophilic granulomatosis with polyangiitis (Churg-Strauss): data on seventeen patients. Arthritis Rheumatol 68:2274–2282
Jandus P, Hausmann O, Haeberli G, Gentinetta T, Mueller U, Helbling A (2011) Unpredicted adverse reaction to omalizumab. J Investig Allergol Clin Immunol 21:563–566
Kavati A, Zhdanava M, Ortiz B, LeCocq J, Schiffman B, Pilon D, Ching Cheung H, Lefebvre P, Stone BD (2019) Long-term omalizumab outcomes in chronic idiopathic urticaria: a real-world study. Allergy Asthma Proc 40:321–328
Klyucheva M, von Berg A, Gappa M, Suerbaum C, Berdel D (2013) Omalizumab therapy in adolescents with severe allergic asthma - results of a post-marketing surveillance. Pneumologie 67:233–237
Kumar SS (2019) Possible omalizumab-induced arthritis. Rheumatology Consultant [published online May 20, 2019]
Lazaridis N, Germanidis G (2018) Current insights into the innate immune system dysfunction in irritable bowel syndrome. Ann Gastroenterol 31:171–187
Lemal R, Fouquet G, Terriou L, Vaes M, Livideanu CB, Frenzel L, Barete S, Canioni D, Lhermitte L, Rossignol J, Arock M, Dubreuil P, Lortholary O, Hermine O (2019) Omalizumab therapy for mast cell-mediator symptoms in patients with ISM, CM, MMAS, and MCAS. J Allergy Clin Immunol Pract 7:2387–2395
Lowe PJ, Tannenbaum S, Gautier A, Jimenez P (2009) Relationship between omalizumab pharmacokinetics, IgE pharmacodynamics and symptoms in patients with severe persistent allergic (IgE-mediated) asthma. Br J Clin Pharmacol 68:61–76
Lyons JJ, Yu X, Hughes JD, Le QT, Jamil A, Bai Y, Ho N, Zhao M, Liu Y, O’Connell MP, Trivedi NN, Nelson C, DiMaggio T, Jones N, Matthews H, Lewis KL, Oler AJ, Carlson RJ, Arkwright PD, Hong C, Agama S, Wilson TM, Tucker S, Zhang Y, McElwee JJ, Pao M, Glover SC, Rothenberg ME, Hohman RJ, Stone KD, Caughey GH, Heller T, Metcalfe DD, Biesecker LG, Schwartz LB, Milner JD (2016) Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat Genet 48:1564–1569
Mariani G, Strober W (1990) Immunoglobulin metabolism. In: Metzger H. Washington DC (eds) American Society of Microbiology Receptors and the Action of Antibodies. pp94–177
Marshall JS, King CA, McCurdy JD (2003) Mast cell cytokine and chemokine responses to bacterial and viral infection. Curr Pharm Des 9:11–24
Molderings GJ, Raithel M, Kratz F, Azemar M, Haenisch B, Harzer S, Homann J (2011) Omalizumab treatment of systemic mast cell activation disease: experiences from four cases. Intern Med 50:611–615
Molderings GJ, Haenisch B, Bogdanow M, Fimmers R, Nöthen MM (2013) Familial occurrence of systemic mast cell activation disease. PLoS One 8:e76241
Molderings GJ, Haenisch B, Brettner S, Homann J, Menzen M, Dumoulin FL, Panse J, Butterfield J, Afrin LB (2016) Pharmacological treatment options for mast cell activation disease. Naunyn Schmiedeberg’s Arch Pharmacol 389:671–694
Molimard M, Mala L, Bourdeix I, Le Gros V (2014) Observational study in severe asthmatic patients after discontinuation of omalizumab for good asthma control. Respir Med 108:571–576
Nopp A, Johansson SG, Ankerst J, Palmqvist M, Oman H (2007) CD-sens and clinical changes during withdrawal of Xolair after 6 years of treatment. Allergy 62:1175–1181
Nopp A, Johansson SG, Adédoyin J, Ankerst J, Palmqvist M, Oman H (2010) After 6 years with Xolair; a 3-year withdrawal follow-up. Allergy 65:56–60
Pabst S, Tiyerili V, Grohe C (2008) Apparent response to anti-IgE therapy in two patients with refractory “forme fruste” of Churg-Strauss syndrome. Thorax 63:747–748
Pavlicek V (2014) Glukokortikoide richtig ausschleichen: warum, wann und wie? Schweiz Med Forum 14:398–401
Pennington LF, Tarchevskaya S, Brigger D, Sathiyamoorthy K, Graham MT, Nadeau KC, Eggel A, Jardetzky TS (2016) Structural basis of omalizumab therapy and omalizumab-mediated IgE exchange. Nat Commun 19:11610–11621
Peterson MR, Coop CA (2017) Long-term omalizumab use in the treatment of exercise-induced anaphylaxis. Allergy Rhinol (Providence) 8:170–172
Pilette C, Coppens N, Houssiau FA, Rodenstein DO (2007) Severe serum sickness-like syndrome after omalizumab therapy for asthma. J Allergy Clin Immunol 120:972–973
Presta LG, Lahr SJ, Shields RL, Porter JP, Gorman CM, Fendly BM, Jardieu PM (1993) Humanization of an antibody directed against IgE. J Immunol 151:2623–2632
Presta L, Shields R, O’Connell L, Lahr S, Porter J, Gorman C, Jardieu P (1994) The binding site on human immunoglobulin E for its high affinity receptor. J Biol Chem 269:26368–26373
Shmagel KV, Chereshnev VA (2009) Molecular bases of immune complex pathology. Biochem Mosc 74:469–479
Quintas-Cardama A, Aribi A, Cortes J, Giles FJ, Kantarjian H, Verstovsek S (2006) Novel approaches in the treatment of systemic mastocytosis. Cancer 107:1429–1439
Rogers AS, Israel E, Smith CR, Levine D, McBean AM, Valente C, Faich G (1988) Physician knowledge, attitudes, and behavior related to reporting adverse drug events. Arch Intern Med 148:1596–1600
Ruppert AM, Averous G, Stanciu D, Deroide N, Riehm S, Poindron V, Pauli G, Debry C, de Blay F (2008) Development of Churg-Strauss syndrome with controlled asthma during omalizumab treatment. J Allergy Clin Immunol 121:253–254
Scott HD, Rosenbaum SE, Waters WJ, Colt AM, Andrews LG, Juergens JP, Faich GA (1987) Rhode Island physicians’ recognition and reporting of adverse drug reactions. R I Med J 70:311–316
Slapnicar C, Trinkaus M, Hicks L, Vadas P (2019) Efficacy of Omalizumab in indolent systemic mastocytosis. Case Rep Hematol 3787586
Starke P (2015) Clinical review of Xolair (omalizumab) - FDA; BLA STN 103976/5149; https://www.fda.gov› media › download
Stokes J (2017) Anti-IgE treatment for disorders other than asthma. Front Med (Lausanne) 4:152
Theoharides TC, Valent P, Akin C (2015) Mast cells, mastocytosis, and related disorders. N Engl J Med 373:163–172
Thomson NC, Chaudhuri R (2012) Omalizumab: clinical use for the management of asthma. Clin Med Insights Circ Respir Pulm Med 6:27–40
Tridente G (2014) Adverse effects with biomedicines: prevention through understanding. Springer, Germany
Valent P, Akin C, Sperr WR, Mayerhofer M, Födinger M, Fritsche-Polanz R, Sotlar K, Escribano L, Arock M, Horny HP, Metcalfe DD (2005) Mastocytosis: pathology, genetics, and current options for therapy. Leuk Lymphoma 46:35–48
Valent P, Akin C, Arock M, Brockow K, Butterfield JH, Carter MC, Castells M, Escribano L, Hartmann K, Lieberman P, Nedoszytko B, Orfao A, Schwartz LB, Sotlar K, Sperr WR, Triggiani M, Valenta R, Horny HP, Metcalfe DD (2012) Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. Int Arch Allergy Immunol 157:215–225
Vichyanond P (2011) Omalizumab in allergic diseases, a recent review. Asian Pac J Allergy Immunol 29:209–219
Weiss SL, Smith DM (2019) A case of serum sickness-like reaction in an adult treated with Omalizumab. Mil Med doi. https://doi.org/10.1093/milmed/usz357
Zampetti A (2018) Mastocytosis- a new therapeutic scenario with omalizumab. Int J Aller Medications 4:027
Zen M, Canova M, Campana C, Bettio S, Nalotto L, Rampudda M, Ramonda R, Iaccarino L, Doria A (2011) The kaleidoscope of glucorticoid effects on immune system. Autoimmun Rev 10:305–310
Acknowledgment
This article is dedicated to Edelgard Klasing, the former chairwoman of Fatigatio e.V., the German patient organization for chronic fatigue syndrome, who died in 2018 due to the interaction described herein.
Funding
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
GJM and FLD conceived and designed the study. All authors contributed data either from their own patients or from research of the literature. GJM analyzed the data. GJM drafted the manuscript which was optimized by comments from all authors. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Gerhard J. Molderings is chief medical officer of the company MC Sciences developing drugs for therapy of mast cell diseases. All other authors have stated that they have no conflicts of interest.
Disclosures
Publication was supported by the Förderclub Mastzellforschung e.V. Informed consents for publication have been obtained from our patients.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Molderings, G.J., Dumoulin, F.L., Homann, J. et al. Adrenal insufficiency is a contraindication for omalizumab therapy in mast cell activation disease: risk for serum sickness. Naunyn-Schmiedeberg's Arch Pharmacol 393, 1573–1580 (2020). https://doi.org/10.1007/s00210-020-01886-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-020-01886-2