
Abstract
The World Health Organization estimates that diabetes prevalence has risen from 108 million in 1980 to 422 million in 2014, with type 2 diabetes accounting for more than 90% of these cases. Furthermore, the prevalence of prediabetes (impaired fasting glucose and/or impaired glucose tolerance) is more than 40% in some countries and is associated with a global rise in obesity. Therefore it is imperative that we develop new approaches to reduce the development of prediabetes and progression to type 2 diabetes. In this review, we explore the gains made over the past decade by focused efforts to improve insulin secretion by the beta cell or insulin sensitivity of target tissues. We also describe multitasking candidates, which could improve both beta cell dysfunction and peripheral insulin sensitivity. Moreover, we highlight provocative findings indicating that additional glucose regulatory tissues, such as the brain, may be key therapeutic targets. Taken together, the promise of these new multi-faceted approaches reinforces the importance of understanding and tackling type 2 diabetes pathogenesis from a multi-tissue perspective.
Similar content being viewed by others


Introduction
According to the International Diabetes Federation, one in every 11 adults worldwide has diabetes, and type 2 diabetes accounts for more than 90% of these cases [1]. Together with the increased prevalence of type 2 diabetes, the rates of prediabetes (defined as impaired fasting glucose and/or impaired glucose tolerance) are booming. Prediabetes represents a transition between normal glucose tolerance and diabetes, and is characterised by milder elevations in the fasting (5.6 to 6.9 mmol/l) and 2 h glucose levels (7.8 to 11.0 mmol/l) in an OGTT, and/or elevated HbA1c (39 to 46 mmol/mol [5.7% to 6.4%]) [2]. The accelerated rates of prediabetes and type 2 diabetes are currently outpacing preventative efforts. Strikingly, the increase in prevalence of dysglycaemia is paralleled by an increase in obesity, with more than 1 in 3 adults classified as overweight and more than 1 in 10 as obese.
In obesity, insulin resistance is manifested by decreased glucose uptake by the insulin-sensitive tissues, resulting in persistent hyperinsulinaemia (Fig. 1a). As the beta cell becomes overburdened in prediabetes, it no longer secretes sufficient insulin, resulting in impaired glycaemia (Fig. 1b). Then, as beta cell function decreases further, type 2 diabetes develops and, with continued disease progression, the beta cell’s insulin response drops even further and glucose levels continue to rise (Fig. 1b). Observations of these phenotypic responses support the concept that chronic hyperglycaemia places a tremendous load on the beta cell, and is likely to play a role in the progression to beta cell failure.

Relationship between beta cell insulin release and peripheral insulin sensitivity in determining states of glycaemic control. Individuals are classified as having normal glucose tolerance, prediabetes or type 2 diabetes based on the evaluation of fasting plasma glucose levels and/or 2 h plasma glucose values after a 75 g OGTT, or HbA1c measurement. With emerging peripheral insulin resistance, beta cells compensate by releasing more insulin (as depicted); (a) in individuals who are not at risk of developing abnormalities of glucose tolerance, the beta cells continue to release more insulin in response to prolonged insulin resistance, and potentially beta cell mass also increases, thereby maintaining normoglycaemia over time. (b) In individuals who are at increased risk of developing diabetes because of genetic or epigenetic susceptibility, beta cells are unable to adequately compensate for emerging peripheral insulin resistance because insulin release is insufficient for the degree of insulin resistance, and mild hyperglycaemia (prediabetes) develops. Over time, the progressive nature of the beta cell defect results in ongoing loss of secretory function and a further decline in beta cell mass such that severe hyperglycaemia (type 2 diabetes) develops. Gluc, glucose; Ins, insulin. This figure is available as part of a downloadable slideset
Because beta cell dysfunction and/or demise has been identified as the critical component responsible for the development of prediabetes and progression to frank type 2 diabetes [3, 4], early research efforts were pointedly focused on beta cell function. Indeed, more recent pharmacological approaches have focused on making the beta cell healthier, to enhance insulin secretion. However, it is uncertain whether requiring a dysfunctional beta cell to work harder is likely to produce more durable glucose control than reducing its workload [5, 6]. Thus, it is important to consider both beta cell dysfunction and peripheral insulin resistance as intervention targets (see the text box). In this review, we summarise the current approaches to treatment of beta cell dysfunction and peripheral insulin resistance. We also describe emerging approaches that target both the beta cells and peripheral insulin target tissues and discuss novel strategies that extend even further beyond the traditional dogma of glycaemic regulation.

Restoring beta cell function
The primary function of the beta cell is to release insulin in response to a rise in blood glucose level. The beta cell of non-diabetic individuals senses nutrients (primarily glucose) within minutes of eating. Upon entry into the beta cell, glucose is rapidly metabolised, increasing the cellular ATP/ADP ratio and triggering the KATP channels at the plasma membrane to close, thus inducing membrane depolarisation and causing the voltage-dependent Ca2+ channels to open. This facilitates the influx of Ca2+ to the cell’s interior, resulting in insulin release; this mechanism is called the triggering pathway of insulin secretion. In human beta cells, there is an amplifying phase that is distinct in dynamics and mechanisms from the triggering phase, producing a biphasic pattern of insulin release [7]. This biphasic insulin release is detectable in response to an intravenous glucose bolus or a step increase in glucose levels in humans, although it is less clear in response to other nutritional stimuli [7]. Biphasic insulin release is also recapitulated by human islets ex vivo in response to a glucose stimulus. A therapy introduced some time ago capitalised on the triggering pathway by activating the KATP/sulfonylurea receptor (SUR) channels using sulfonylureas to stimulate insulin release (Fig. 2). While effective at initially reducing hyperglycaemia, these agents may ‘push’ the beta cell too much, and hasten beta cell exhaustion and death [8, 9].

Beta cell function, peripheral insulin sensitivity and points of entry for therapeutic targeting. In the islet beta cell (blue), glucose enters via the GLUT1/2 glucose transporter. Its intracellular metabolism increases the ATP/ADP ratio, triggering closure of KATP channels, stimulating plasma membrane depolarisation (Ψ) and opening of the voltage-dependent calcium channels, thereby permitting entry of extracellular Ca2+ into the cell. The net increase in intracellular Ca2+ facilitates SNARE complex-regulated GSIS. GLP-1 binds to the GLP-1 receptor, and increases cAMP and amplifies GSIS. Sulfonylureas stimulate insulin release by binding to and closing (thus activating) KATP/SUR channels. In the skeletal muscle cell (pink), circulating insulin binds to the insulin receptor to trigger canonical insulin signalling through IRS-PI3K. This leads to F-actin remodelling to provide tracks upon which GLUT4 vesicles travel to SNARE proteins at the plasma membrane for subsequent docking and fusion to facilitate glucose uptake. Factors that multitask in both beta cell- and muscle-specific processes and act positively include STX4, PAK1, BMP7 and osteocalcin, while TXNIP exerts negative actions. Dashed lines indicate pathways that are as yet unclear. GLP-1R, GLP-1 receptor; IR, insulin receptor; SNARE, SNAP (soluble NSF [N-ethylmaleimide-sensitive factor] attachment protein) receptor; VDCC, voltage-dependent calcium channel. This figure is available as part of a downloadable slideset
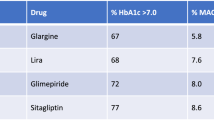
Over the past decade or so, an alternative approach to improving beta cell function has been to amplify insulin release in a glucose-dependent manner by enhancing the action of the incretin peptides. Glucagon-like peptide 1 (GLP-1) receptor agonists are effective in promoting biphasic insulin release (Fig. 2). They also rarely cause hypoglycaemia or body weight gain—a step forward in type 2 diabetes treatment. Another approach to enhancing beta cell function is the use of dipeptidyl peptidase 4 (DPP-4) inhibitors, which prolong the half-life of incretins, such as GLP-1, by preventing their rapid degradation [10]. Stemming from this are advances in the delivery of GLP-1/glucagon-derived peptides [11]. In preclinical studies, such peptides capitalise upon the existence of multiple receptor targets; for example, these peptides may act through glucagon, GLP-1 and glucose-dependent insulinotropic polypeptide (GIP) receptors to reduce body weight and enhance glycaemic control [11]. However, the HbA1c profile is similar between these newer agents and sulfonylureas; thus, these approaches, which all increase the workload for a dysfunctional beta cell, may not be as successful at maintaining glucose control as had been initially hoped [5, 12]. Indeed, reducing the workload for a dysfunctional beta cell is more likely to produce durable glucose control [6, 13]. In support of this concept, the ADOPT (A Diabetes Outcome Progression Trial) study demonstrated that promoting insulin sensitisation is a viable approach to reduce beta cell workload and remedy glucose control [9].
Clinical approaches to insulin sensitisation
Lifestyle intervention (weight loss via exercise and diet) and medications improve insulin sensitivity and enhance beta cell function [4, 9]. Therefore, efforts to reduce insulin resistance and thus the beta cell’s burden have been shown to be effective in preventing progression of prediabetes to type 2 diabetes and worsening of diabetes. Thiazolidinediones efficiently mitigate a portion of the insulin resistance associated with type 2 diabetes. Although they were widely used in the 1990s and 2000s, their adverse effects (weight gain, oedema, bone fractures) were felt by many to outweigh their benefits and now they are hardly used. Metformin, a widely used first-line therapy that ameliorates hyperglycaemia by decreasing hepatic glucose output, is often insufficient on its own, over the longer term [8]. More recently, the sodium–glucose cotransporter-2 (SGLT2) inhibitors were introduced as a workaround for peripheral insulin resistance. These agents induce glycosuria, thereby reducing the need for insulin to dispose of glucose. Studies in animals and humans suggest that this approach, by reducing chronic hyperglycaemia, may improve insulin sensitivity and beta cell function [14]. However, it remains unknown whether these agents can maintain long-term glucose control.
Recent studies have shown that bariatric surgery rapidly improves beta cell function, preceding any notable change in obesity or adiposity, suggesting that the surgery triggers the release of factors that benefit beta cell function. However, the mechanisms by which this might occur have not as yet been definitively identified. Mechanisms involving gut hormones (e.g. GLP-1, GIP), gut microbiota, bile acids, fibroblast growth factor (FGF) 19 and improved hepatic or skeletal muscle insulin sensitivity, are all active postulates under investigation [15].
Multitasking factors in diabetes therapy
An alternative to targeting either beta cell dysfunction or insulin resistance is to target single factors that multitask in beta cells and peripheral insulin-sensitive cells. These multitasking factors enhance the efficiency of glucose-stimulated insulin secretion (GSIS) and insulin-stimulated glucose uptake, respectively, in a coordinated fashion. Current research efforts are focused on endogenous factors that multitask in beta cells and insulin-sensitive cells and show promise in preclinical studies and ex vivo human islet studies. For example, type 2 diabetic human islets are deficient in the exocytosis factor syntaxin 4 (STX4), and replenishing it restores their function—STX4 enrichment protects beta cell function against diabetogenic stimuli (e.g. obesity, glucolipotoxicity), while also promoting peripheral insulin sensitivity [16, 17] (Fig. 2). Another multitasking factor, p21-activated kinase 1 (PAK1), is a key mediator of stimulus-induced actin remodelling and is deficient in type 2 diabetic human islets. Enrichment of PAK1 protects beta cell function and supports skeletal muscle cell glucose uptake [18, 19]. Similarly, restoration of bone morphogenic factor 7 (BMP7) deficiency in mouse models of prediabetes/diabetes largely resolves hyperglycaemia via improved skeletal muscle insulin sensitivity [20]. BMP7, a member of the TGF-β-superfamily, confers glucose-sensitive insulin release to beta cell progenitors [21], although the impact of BMP7 on pancreatic islet function in vivo remains to be evaluated. Moreover, the bone-derived factor osteocalcin also promotes GSIS in beta cells [22] and enhances skeletal muscle glucose uptake [23]. Although most of the identified multitasking factors are deficient in type 2 diabetes, some are overexpressed and may contribute to diabetes progression. For instance, thioredoxin interacting protein (TXNIP) expression is elevated in type 2 diabetic human skeletal muscle, and its silencing confers improved peripheral tissue glucose uptake [24]; TXNIP inhibition also indirectly promotes beta cell function [25]. Clearly, further work is needed to determine whether any of these candidates will have applicability to the treatment of humans.
New strategies for diabetes therapy
The combined roles of defects in beta cell function and peripheral insulin sensitivity in type 2 diabetes are well established. However, provocative new studies suggest that the opportunities for therapeutic control of insulin sensitivity extend even further beyond this signalling network. For instance, FGF1 is a multifunctional growth factor that activates all FGF receptor subtypes and is present on both beta cells and peripheral insulin-sensitive tissues. In preclinical studies, when FGF1 was delivered via intracerebroventricular injection rather than peripherally, it resolved diabetes following a single injection without the development of either hypoglycaemia or obesity [26]. It is particularly notable that the intracerebroventricular mode of delivery confers a therapeutic advantage, supporting a recent surge in research investigating how the central nervous system influences islet function and peripheral insulin sensitivity to orchestrate glucose homeostasis [27].
In accordance with the goals of precision medicine, diabetes treatment strategies could also benefit from a more refined assessment of the patient phenotype. We have seen this in the identification of neonatal diabetes and MODY genotypes [28, 29]. Recently, a study using cluster analysis suggested that optimal treatment strategies and target tissues could differ based upon how individuals with type 2 diabetes cluster phenotypically. This assessment used an analysis of six variables [30], as opposed to the one variable typically used to define prediabetes and type 2 diabetes—glycaemia. Although this study covers populations predominantly from Northern Europe and needs to be reproduced elsewhere, it reveals possibilities that may impact on the choice of glucose-lowering therapies, allowing more nuanced treatment strategies tailored to the particular cluster-type. We believe this observation now requires rigorous replication in other populations to allow us to determine whether we should rethink how we categorise diabetes that is not immune in nature.
Identifying new therapeutic targets via genetics and epigenetics
In addition to the targets already the subject of preclinical studies, a growing list of new therapeutic candidates is emerging from genomic studies. An early type 2 diabetes genome-wide association study (GWAS) pointed to 15 genes, 33% with SNPs encoding factors involved in beta cell function and 9% with linkages to insulin action [31]. Subsequent studies spanning more than 10 years culminated in relatively similar findings. These observations supported a focus on improving beta cell function and insulin sensitisation as approaches to combat prediabetes and type 2 diabetes. However, one concern is that current approaches may be missing rare variants; rare coding mutations in the genes located near the most associated SNPs can establish causality beyond the GWAS method. In the new era of precision medicine, this search for rare variants has been proposed as an alternative means to identify novel targets for future therapies, potentially filling a gap in ‘missing type 2 diabetes heritability’ [32]. A broad example of the efficacy of this approach has been the successful identification of type 2 diabetic carriers of specific monogenic diabetes (MODY) mutations who responded better to sulfonylureas than to insulin [28]. However, the approach is limited by issues such as penetrance [33], or confounded by conflicting preclinical functional data (reviewed in [34]).
Missing type 2 diabetes heritability may also be linked to a role for epigenetic DNA modifications and non-coding RNAs as key players in the pathogenesis of type 2 diabetes. Epigenetic DNA modifications, such as DNA methylation and histone acetylation, are strictly regulated to maintain optimal tissue-specific gene expression profiles. However, epigenetic modifications can be altered based on environmental cues, such as exercise, diet and the intrauterine environment, which can modify the risk for type 2 diabetes. For example, altered DNA methylation patterns have been reported for functionally important genes in islets, skeletal muscle and adipose tissues in type 2 diabetic vs non-diabetic donors [35,36,37]. Moreover, it was recently shown that ‘metabolic memory’ is conferred in epigenetic changes due to hyperglycaemia [38]. DNA methylation, coupled with genetic variation analysis, has recently been used to determine that 50% of known type 2 diabetes SNPs are associated with altered DNA methylation. One locus (KCNQ1) was found for which methylation predicts a causal pathway to type 2 diabetes, as opposed to being the result of disease [39]. Post-transcriptional gene silencing also responds to microRNAs (miRNAs), which are 20–25 nucleotide non-coding RNAs. One miRNA can influence the expression of several targets, or conversely, several miRNAs can regulate expression of a single gene. miRNAs regulate critical components of GSIS in the beta cell [40, 41] and also skeletal muscle mitochondrial biogenesis and insulin signalling by targeting genes such as PI3K and GLUT4 (also known as SLC2A4) [42]. Inhibition of miRNA-103 and miRNA-107 has been shown to significantly enhance insulin sensitivity [43]. Long non-coding RNAs (lncRNAs), which are more than 200 nucleotides long, are also emerging as important factors in type 2 diabetes. BetaLinc1 (beta cell long intergenic non-coding RNA 1) has been shown to be important for islet beta cell formation and function in mice [44]. Hence, exploiting miRNA and lncRNA targets is an active area of type 2 diabetes research, although it remains unknown whether there are miRNA and lncRNA targets that can multitask to address beta cell dysfunction and insulin resistance in a coordinated fashion.
Small extracellular vesicles, often called exosomes (50–150 nm in diameter), carry miRNAs, other nucleic acids and proteins. They are secreted by cells and could be involved in cell-to-cell communication and inter-organ crosstalk in beta cells and insulin-responsive tissues [45]. In humans physical exercise significantly enhances release of exosomes into the circulation [46]. While most mechanistic data captured to date are preclinical, provocative data from high fat diet-fed mice suggest that exosomes derived from skeletal muscle modulate the gene expression and proliferation rates of clonal beta cells, and that miR-16 is a key signalling factor in the exosomes [45]. Moreover, adipose tissue macrophage-derived exosomes containing miRNAs from obese mice caused insulin resistance in lean mice; this effect was attributed to increased expression of miR-155 in adipose tissue macrophages from the obese mice [47]. Conversely, exosomes from lean mice improved glucose tolerance in obese mice [47]. Harnessing the potential of cell–cell communication by exosomes could represent a new delivery tool for therapeutic agents.
Conclusions and perspectives
This review highlights the importance of understanding type 2 diabetes pathogenesis from a multi-tissue angle and points out that strategies focused on improving insulin sensitivity could be crucial for beta cell health in the treatment of type 2 diabetes. Conventional medications are largely insufficient to attain long-term remission of type 2 diabetes, with some commonly causing unwanted effects such as weight gain and hypoglycaemia. In reassessing the progression from prediabetes to type 2 diabetes (Fig. 1), and considering frank type 2 diabetes itself, one has to question whether our approach of pushing a dysfunctional beta cell to make and release more insulin without co-resolution of insulin resistance is hastening beta cell failure and disease progression. Thus, is it not time to think more broadly about the prevention and treatment of type 2 diabetes?
Abbreviations
- ADOPT:
-
A Diabetes Outcome Progression Trial
- BMP7:
-
Bone morphogenic factor 7
- DPP-4:
-
Dipeptidyl peptidase 4
- FGF:
-
Fibroblast growth factor
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- GLP-1:
-
Glucagon-like peptide 1
- GSIS:
-
Glucose-stimulated insulin secretion
- GWAS:
-
Genome-wide association study
- lncRNA:
-
Long non-coding RNA
- miRNA:
-
MicroRNA
- PAK1:
-
p21-activated kinase 1
- PI3K:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- STX4:
-
Syntaxin 4
- SUR:
-
Sulfonylurea receptor
- TXNIP:
-
Thioredoxin interacting protein
References
International Diabetes Federation (2017) Diabetes Atlas, 8th edn. Available from http://www.diabetesatlas.org/. Accessed 13 Nov 2017
American Diabetes Association (2018) 2. Classification and diagnosis of diabetes: standards of medical care in diabetes—2018. Diabetes Care 41(Suppl 1):S13–S27
Weyer C, Bogardus C, Mott DM, Pratley RE (1999) The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest 104:787–794
Kitabchi AE, Temprosa M, Knowler WC et al (2005) Role of insulin secretion and sensitivity in the evolution of type 2 diabetes in the diabetes prevention program: effects of lifestyle intervention and metformin. Diabetes 54:2404–2414
Nauck M, Frid A, Hermansen K et al (2009) Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care 32:84–90
Cefalu WT, Leiter LA, Yoon KH et al (2013) Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet 382:941–950
Henquin JC, Dufrane D, Gmyr V, Kerr-Conte J, Nenquin M (2017) Pharmacological approach to understanding the control of insulin secretion in human islets. Diabetes Obes Metab 19:1061–1070
Kahn SE, Haffner SM, Heise MA et al (2006) Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med 355:2427–2443
Kahn SE, Lachin JM, Zinman B et al (2011) Effects of rosiglitazone, glyburide, and metformin on β-cell function and insulin sensitivity in ADOPT. Diabetes 60:1552–1560
McIntosh CH, Demuth HU, Pospisilik JA, Pederson R (2005) Dipeptidyl peptidase IV inhibitors: how do they work as new antidiabetic agents? Regul Pept 128:159–165
Finan B, Yang B, Ottaway N et al (2015) A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med 21:27–36
Seck T, Nauck M, Sheng D et al (2010) Safety and efficacy of treatment with sitagliptin or glipizide in patients with type 2 diabetes inadequately controlled on metformin: a 2-year study. Int J Clin Pract 64:562–576
DeFronzo RA, Tripathy D, Schwenke DC et al (2011) Pioglitazone for diabetes prevention in impaired glucose tolerance. N Engl J Med 364:1104–1115
Ferrannini E, Muscelli E, Frascerra S et al (2014) Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest 124:499–508
Mulla CM, Middelbeek RJW, Patti ME (2018) Mechanisms of weight loss and improved metabolism following bariatric surgery. Ann N Y Acad Sci 1411:53–64
Oh E, Stull ND, Mirmira RG, Thurmond DC (2014) Syntaxin 4 up-regulation increases efficiency of insulin release in pancreatic islets from humans with and without type 2 diabetes mellitus. J Clin Endocrinol Metab 99:E866–E870
Oh E, Miller RA, Thurmond DC (2015) Syntaxin 4 overexpression ameliorates effects of aging and high-fat diet on glucose control and extends lifespan. Cell Metab 22:499–507
Ahn M, Yoder SM, Wang Z et al (2016) The p21-activated kinase (PAK1) is involved in diet-induced beta cell mass expansion and survival in mice and human islets. Diabetologia 59:2145–2155
Tunduguru R, Chiu TT, Ramalingam L, Elmendorf JS, Klip A, Thurmond DC (2014) Signaling of the p21-activated kinase (PAK1) coordinates insulin-stimulated actin remodeling and glucose uptake in skeletal muscle cells. Biochem Pharmacol 92:380–388
Chattopadhyay T, Singh RR, Gupta S, Surolia A (2017) Bone morphogenetic protein-7 (BMP-7) augments insulin sensitivity in mice with type II diabetes mellitus by potentiating PI3K/AKT pathway. Biofactors 43:195–209
Klein D, Alvarez-Cubela S, Lanzoni G et al (2015) BMP-7 induces adult human pancreatic exocrine-to-endocrine conversion. Diabetes 64:4123–4134
Ferron M, Hinoi E, Karsenty G, Ducy P (2008) Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci U S A 105:5266–5270
Zhou B, Li H, Xu L, Zang W, Wu S, Sun H (2013) Osteocalcin reverses endoplasmic reticulum stress and improves impaired insulin sensitivity secondary to diet-induced obesity through nuclear factor-κB signaling pathway. Endocrinology 154:1055–1068
Parikh H, Carlsson E, Chutkow WA et al (2007) TXNIP regulates peripheral glucose metabolism in humans. PLoS Med 4:e158
Xu G, Chen J, Jing G, Shalev A (2013) Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat Med 19:1141–1146
Scarlett JM, Rojas JM, Matsen ME et al (2016) Central injection of fibroblast growth factor 1 induces sustained remission of diabetic hyperglycemia in rodents. Nat Med 22:800–806
Ruud J, Steculorum SM, Bruning JC (2017) Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat Commun 8:15259
Shepherd M, Pearson ER, Houghton J, Salt G, Ellard S, Hattersley AT (2003) No deterioration in glycemic control in HNF-1α maturity-onset diabetes of the young following transfer from long-term insulin to sulphonylureas. Diabetes Care 26:3191–3192
Pearson ER, Flechtner I, Njolstad PR et al (2006) Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 355:467–477
Ahlqvist E, Storm P, Karajamaki A et al (2018) Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol 6:361–369
Barroso I, Luan J, Middelberg RP et al (2003) Candidate gene association study in type 2 diabetes indicates a role for genes involved in beta-cell function as well as insulin action. PLoS Biol 1:E20
Lawlor N, Khetan S, Ucar D, Stitzel ML (2017) Genomics of islet (dys)function and type 2 diabetes. Trends Genet 33:244–255
Majithia AR, Flannick J, Shahinian P et al (2014) Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc Natl Acad Sci U S A 111:13127–13132
Davidson HW, Wenzlau JM, O'Brien RM (2014) Zinc transporter 8 (ZnT8) and beta cell function. Trends Endocrinol Metab 25:415–424
Dayeh T, Volkov P, Salo S et al (2014) Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet 10:e1004160
Nitert MD, Dayeh T, Volkov P et al (2012) Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes 61:3322–3332
Nilsson E, Jansson PA, Perfilyev A et al (2014) Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 63:2962–2976
Chen Z, Miao F, Paterson AD et al (2016) Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc Natl Acad Sci U S A 113:E3002–E3011
Elliott HR, Shihab HA, Lockett GA et al (2017) Role of DNA methylation in type 2 diabetes etiology: using genotype as a causal anchor. Diabetes 66:1713–1722
Ofori JK, Salunkhe VA, Bagge A et al (2017) Elevated miR-130a/miR130b/miR-152 expression reduces intracellular ATP levels in the pancreatic beta cell. Sci Rep 7:44986
Poy MN, Eliasson L, Krutzfeldt J et al (2004) A pancreatic islet-specific microRNA regulates insulin secretion. Nature 432:226–230
Zhou T, Meng X, Che H et al (2016) Regulation of insulin resistance by multiple MiRNAs via targeting the GLUT4 signalling pathway. Cell Physiol Biochem 38:2063–2078
Trajkovski M, Hausser J, Soutschek J et al (2011) MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 474:649–653
Arnes L, Akerman I, Balderes DA, Ferrer J, Sussel L (2016) βlinc1 encodes a long noncoding RNA that regulates islet β-cell formation and function. Genes Dev 30:502–507
Jalabert A, Vial G, Guay C et al (2016) Exosome-like vesicles released from lipid-induced insulin-resistant muscles modulate gene expression and proliferation of beta recipient cells in mice. Diabetologia 59:1049–1058
Fruhbeis C, Helmig S, Tug S, Simon P, Kramer-Albers EM (2015) Physical exercise induces rapid release of small extracellular vesicles into the circulation. J Extracell Vesicles 4:28239
Ying W, Riopel M, Bandyopadhyay G et al (2017) Adipose tissue macrophage-derived exosomal miRNAs can modulate in vivo and in vitro insulin sensitivity. Cell 171:372–384.e12
Acknowledgements
The authors apologise to all those whose contributions to the field could not be cited because of space limitations. We thank N. Linford (Linford Biomedical Communications, LLC, Spokane, WA, USA) for editing assistance.
Funding
Work in the authors’ laboratories is supported by grants from the National Institutes of Health (DK067912 and DK102233 to DCT; P30DK017047 to SEK) and the Department of Veterans Affairs (I01BX001060 to SEK).
Author information
Authors and Affiliations
Contributions
VAS, RV, SEK and DCT conceived of and drafted the manuscript and approved its final version. DCT is responsible for the integrity of this work. All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Corresponding author
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Vishal A. Salunkhe and Rajakrishnan Veluthakal are joint first authors of this review.
Electronic supplementary material
ESM Downloadable slideset
(PPTX 428 kb)
Rights and permissions
About this article

Cite this article
Salunkhe, V.A., Veluthakal, R., Kahn, S.E. et al. Novel approaches to restore beta cell function in prediabetes and type 2 diabetes. Diabetologia 61, 1895–1901 (2018). https://doi.org/10.1007/s00125-018-4658-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-018-4658-3