
Abstract
Aims/hypothesis
We investigated the effect of elevated circulating NEFA on insulin-mediated hepatic glucose uptake (HGU) and whole-body glucose disposal (M) in eight healthy male subjects.
Methods
Studies were performed using positron emission tomography (PET) and [18F]-2-fluoro-2-deoxyglucose ([18F]FDG) during euglycaemic hyperinsulinaemia (0–120 min) and an Intralipid/heparin infusion (IL/Hep; −90–120 min). On a different day, similar measurements were taken during euglycaemic hyperinsulinaemia and saline infusion (SAL). Graphical and compartmental analyses were used to model liver data.
Results
Circulating NEFA increased approximately three-fold during IL/Hep, and declined by 81±7% in the SAL study (p≤0.01). Both M (−28±7%) and HGU (−25±9%) were significantly lowered by NEFA elevation (p=0.004 and p=0.035 respectively). In the whole data set, the decreases in M and HGU were positively correlated (r=0.78, p=0.038). No evidence of [18F]FDG outflow was detected during the scanning time. HGU was correlated with the phosphorylation rate parameter (r=0.71, p=0.003) as derived by compartmental modelling.
Conclusions/interpretation
In healthy men, NEFA impair insulin-mediated HGU and whole-body glucose uptake to a similar extent. Our data suggest that multiple intracellular NEFA targets may concur to down-regulate glucose uptake by the liver.
Similar content being viewed by others
Introduction
Hepatic glucose uptake (HGU) is a determinant of plasma glucose levels [1, 2] and its regulation has received increasing attention. Several studies [2, 3, 4, 5, 6], including most recent dose–response experiments [7, 8], have shown insulin to increase HGU. The ability of insulin to stimulate HGU is impaired in patients with Type 2 diabetes [9, 10, 11], in whom a defect in a proximal step of liver glucose uptake has been demonstrated [9, 10, 12].
High circulating NEFA, commonly observed in patients with Type 2 diabetes [13, 14] impair insulin-mediated glucose disposal in the heart and skeletal muscle [14, 15]. Due to its anatomical location, the liver is highly exposed to the effects of NEFA, since visceral fat directly drains NEFA to the organ through the portal vein. Demonstration of a cause–effect relationship between circulating NEFA and HGU in humans is lacking. Indirect evidence includes data in healthy and insulin-resistant subjects [11] and in patients with Type 2 diabetes [11, 16], showing that HGU is correlated with the ability of insulin to suppress NEFA concentrations. In studies on dogs, HGU was reduced by NEFA elevation [17, 18]. In humans, the administration of the antilipolytic drug, nicotinic acid, produced a significant enhancement in hepatic glucose effectiveness [19].
During insulin plus Intralipid/heparin infusion, glucose uptake in the entire splanchnic bed (splanchnic glucose uptake, SGU) has been found to be reduced in patients with Type 2 diabetes [20], but was either unchanged [21] or increased [22] in healthy individuals. However, glucose disposal by the splanchnic area is predominantly accounted for by extra-hepatic organs, especially during euglycaemia [23]. Using the uridine diphosphate glucose technique, a mild increase in plasma NEFA levels did not alter the incorporation of extracellular glucose into glycogen in healthy subjects [21]. As this observation was made under hyperglycaemic conditions, it is possible that a defect in insulin action may have been masked by the mass action effect of hyperglycaemia on HGU.
Direct effects of NEFA on glucokinase and glucose-6-phosphatase transcription or activity, as well as on hepatic glycogen stores have been described [24, 25, 26, 27, 28], and high NEFA fluxes have been associated with an impairment of hepatic insulin clearance in animal studies [29]. Whether NEFA decrease liver glucose uptake in humans is unknown.
The present study was undertaken to investigate the effect of elevated circulating NEFA on insulin-mediated HGU and whole-body glucose disposal in humans. Healthy individuals were studied under euglycaemic hyperinsulinaemic conditions with and without a concomitant elevation of circulating NEFA. Positron emission tomography (PET) was used to isolate the liver from the other splanchnic organs, in combination with either graphical analysis or kinetic modelling for the calculation of HGU and for information on the early steps involved in tracer uptake and release.
Subjects and methods
Subjects
A total of 16 studies were performed in eight male subjects (age 27±2 years; BMI 22.9±0.7 kg/m2). Each volunteer participated in two studies performed in random order within 1 week. All were healthy as judged by history, physical examination (blood pressure 114±7/62±9 mm Hg; heart rate 63±4 bpm) and routine laboratory tests. None of the subjects were taking any medication and all were instructed to consume a weight-maintaining diet containing at least 200 g carbohydrate per day for 3 days prior to each study. The study protocol was approved by the ethics committee of Turku University Central Hospital, Finland. Each subject gave written informed consent before taking part in the study.
Study design
All studies were conducted after a 10–12-hour overnight fast. Studies consisted of a 90-min baseline period followed by 120 min of euglycaemic hyperinsulinaemia. On the first study day, serum NEFA concentrations were increased by an i.v. administration of Intralipid (IL; 20%; Kabi Vitrum, Uppsala, Sweden) and heparin (Hep; Organon, Helsinki, Finland). Rates of IL and Hep continuous infusion were 60 ml/h and 15 U/min respectively for the entire duration of the study. In addition, two 200-U boluses of heparin were administered at −90 and 0 min respectively. On the second study day, an infusion of saline (SAL; 0.9% NaCl) was given instead of IL/Hep during similar baseline and hyperinsulinaemic periods. For measurement of HGU, [18F]-2-fluoro-2-deoxyglucose ([18F]FDG) was injected after 80±4 min of hyperinsulinaemia, and a 40-min dynamic PET scan was carried out.
PET scanning
Scans were performed using an ECAT 931-08/12 scanner (CTI, Knoxville, Tenn., USA) with a 10.5-cm axial field of view and a resolution of 6.7 mm (axial) × 6.5 mm (in-plane) full width at half maximum. After optimisation of subject position, a transmission scan was performed with a removable 68Ge ring source to correct all subsequent emission data for tissue attenuation of gamma photons. Two intravenous catheters were inserted, one into an antecubital vein for infusion of SAL or IL/Hep, insulin and glucose, as well as for [18F]FDG injection, and into a contralateral heated vein for sampling of arterialised venous blood. After 90 min had elapsed from the start of the IL/Hep or SAL infusion, a primed continuous (40 mU·m−2·min−1) infusion of insulin was started, and a 120-min euglycaemic–hyperinsulinaemic clamp was carried out as previously described [30]. Euglycaemia was maintained using a 20% glucose infusion, the rate of which was adjusted based on frequent plasma glucose measurements. After 80±4 min of euglycaemic hyperinsulinaemia, [18F]FDG (~220 MBq; radiochemical purity >97–99%; specific activity >75 GBq·µmol−1 [31]) was infused intravenously over 30–60 s and a 40-min dynamic scan was carried out to image the liver and cardiac regions, as described previously [15]. Arterialised blood samples were collected throughout scanning to measure whole-blood and plasma [18F]FDG radioactivity over time.
Image processing
All sinograms were corrected for tissue attenuation, dead time and decay, and were reconstructed through standard reconstruction algorithms in a 256×256 matrix. Final in-plane resolution of reconstructed and Hann-filtered images was ~10 mm at 10 cm from the centre of the gantry. Large circular regions of interest (ROI) for hepatic [18F]FDG time–activity measurements were placed on two to four consecutive image planes in the right lobe of the liver. Tissue radioactivity over time was weighted in proportion to the duration of each frame. Smaller ROI were drawn on two adjacent image planes in the left ventricular chamber of the heart for the measurement of radioactivity in arterial blood. Special attention was paid to avoid contamination by surrounding myocardial tissue, and the later points of the curve were corrected for spillover using in vitro measurements of arterialised plasma radioactivity. Input functions were corrected for time delay by comparing them with corresponding tissue time–activity curves [32]. The input function represents the amount of tracer that is available to the organ over time. Though the administered tracer dose and its specific activity are meant to be similar in all studies, the use of an input function ensures that even minimal interstudy differences in circulating tracer levels have no effect on the resulting estimates.
Data analysis
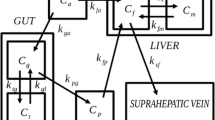
Plasma (input function) and tissue time–activity curves were analysed graphically [33] to quantify the fractional rate of tracer transport and phosphorylation, reflecting glucose influx rate constant into the liver (HKi, ml·min−1·ml−1, or min−1). Graphical analysis has previously been applied to in vivo liver [18F]FDG influx determination in both animal and human studies [6, 11, 16, 34, 35, 36]. As shown in Figure 1, graphical analysis assumes no fixed compartmental configuration, with the tracer moving bidirectionally between plasma and an unspecified number of reversible compartments, and unidirectionally from the latter to an irreversible compartment. With the intrinsic assumptions of tracer theory (i.e. the tracer is derived from plasma; it does not perturb the equilibrium of the measured process; it is transferred by first-order kinetics with rate constant Ki; and it is not initially present in the system), graphical analysis is best suited to steady-state conditions of the measured process (hepatic glucose uptake in this case), and changing concentrations of the injected tracer. In this model, a graph is generated by plotting the following:

a. Graphical analysis model showing tracer moving bidirectionally between plasma (Cp) and an undetermined number (n) of reversible compartments (Ce1-n), and to an irreversible region (Cm). Movement of tracer out of the ‘irreversible’ region back to plasma in the modified model, which accounts for tracer escape from the irreversible compartment, is represented by kloss. b. Plot of measured tracer concentrations in plasma (Cp) and tissue (Ct) over time according to graphical analysis formulae and goodness of fit, as expressed in terms of the r value of the line fitted to the measured points, the slope of which represents the influx rate constant, Ki (ml·min−1·ml−1). HKi=0.0042; r=0.99. c. Three-compartmental model and rate constants (k) showing a fixed compartmental configuration in which the tracer moves bidirectionally between plasma and two tissue compartments. Given large fenestrations in sinusoidal space and low-affinity transport, the effects of blood flow and bidirectional transport are combined as one compartmental exchange (K1, k2), whereas the rate-limiting steps, i.e. phosphorylation (k3) and dephosphorylation rate (k4) regulate movement of tracer across a separate compartment. d. Plot of measured tracer concentrations in tissue over time (open squares) and their prediction by the model (fitted curve, open circles), showing an optimal approximation (goodness of fit), where the sum of squares represents the statistical comparison criterion
where Ct is tissue radioactivity at each sampling time point (t) and Cp is plasma radioactivity. When tracer influx occurs, the two variables describe a linear relationship after a few minutes of equilibration, provided that an irreversible compartment is present. The influx rate constant (HKi) is then given by the slope of the linear fit of the data, after excluding the first few values, as shown in Figure 1. Rate constant values were multiplied by the steady-state plasma glucose concentrations achieved during the clamp to derive net HGU (µmol·min−1·ml−1)[37], given that the relative affinities of glucose and FDG for liver glucose transporters and glucokinase are very close to unity, as shown by in vitro and in vivo experiments [34, 38].
Based on previous results [6, 39], the current scan duration was selected to avoid the confounding effect of tracer outflow from liver cells, and to solely target HGU. The following three independent approaches, which are commonly used to detect significant tracer release from tissue [40, 41], were implemented here to confirm this assumption:
-
1.
To evaluate linearity of the measured data in graphical analysis, the goodness of the linear fit (r value) was obtained in each study, and graphical analysis was repeated over progressive time intervals by changing the starting time of linear fitting. The occurrence of tracer outflow with this method would be revealed by a time-dependent decrease in the calculated slope (HKi) and HGU [41]. Graphical analysis has the advantage of being independent of compartment number and configuration, and thus of the tissue to be targeted [40]. It also verifies (or rejects) the assumption that an irreversible compartment is present within the biological model under study.
-
2.
A modification to the original graphical model was introduced by the authors in order to account for reversible tracer uptake [40]. In this case, it is assumed that there can be loss of tracer from the ‘irreversible’ region back to plasma. Consequently, the change in the amount of tracer in the tissue compartment, dCt(t)/dt is given by the difference between the original tracer inflow and the product of kloss×dCt(t)/dt. Using this method, a rate constant parameter accounting for outflow, referred to here as kloss, is added to the linear model, and kloss-adjusted HKi and HGU can be recalculated accordingly. In this case, the graph is generated by plotting the following:
-
3.
A three-compartment, i.e. plasma [18F]FDG, tissue [18F]FDG and tissue [18F]FDG-6-P, four-rate constant (4k) kinetic model was used, in which the rate constants k3 (min–1) and k4 (min–1) reflect the phosphorylation and dephosphorylation rates respectively [6, 41, 42] (Fig. 1). The differential equations describing the three-compartmental [18F]FDG kinetic model in the liver have been described elsewhere [6, 42] and are given here in Figure 1. In this model, an increase in k4 would suggest that a higher fraction of intracellular tracer undergoes dephosphorylation, resulting in either release or recycling, during the course of the current scanning time [41]. Model rate constants and fractional blood volume were estimated by non-linear least-squares method, minimising the sum of squares (SS) between model-predicted and measured data. SS scores were reported (only) to compare the adequacy of the model between IL/Hep and SAL experiments.
Whole-body glucose uptake (M) was calculated during the steady-state phase of the clamp.
Analytical procedures
Plasma glucose was measured in duplicate by the glucose oxidase technique (Glucose Analyzer II; Beckman Instruments, Fullerton, Calif., USA). Plasma insulin was quantified by radioimmunoassay, and serum NEFA were measured fluorimetrically.
Statistical analysis
All data are presented as means ± SE. Differences in paired data were evaluated using the Student’s paired t test for single repeated measurements. One-way ANOVA was used for unpaired comparisons. Regression analysis was carried out by standard techniques. A p value of less than 0.05 was considered significant.
Results
The metabolic characteristics of the study group during the two experiments are shown in Table 1. Fasting plasma glucose and insulin levels were similar in the SAL and IL/Hep studies. An approximately three-fold increase in circulating NEFA was gradually achieved during the 90-min baseline period of the IL/Hep study, and maintained throughout the clamp (Fig. 2). Conversely, in control SAL studies, NEFA declined by 81±7 % (p≤0.01 vs fasting and vs IL/Hep). Euglycaemia was maintained throughout the clamp on both occasions (NS vs fasting values). Plasma insulin levels were significantly increased during the clamp, and similar during the two experiments.

Circulating concentrations of NEFA during the IL/Hep study (filled circles) and the SAL study (open circles). ** p<0.01 vs baseline; § p<0.01 vs SAL
Optimal image quality was obtained in 15 out of 16 PET studies. In one study, ROI could not be drawn reliably, and this subject was excluded from pairwise HGU analyses. Both M and HGU were significantly lowered by NEFA elevation (Fig. 3a, b). In terms of percentage, the magnitude of the decline was comparable in the two insulin-mediated processes (−28±7 and −25±9% change respectively; NS). An example of graphical plots in the two experiments is shown in Figure 4, in which the slope of the fitted line equals HKi, and is used to calculate HGU. Changes in M and HGU were positively correlated with one another in the entire data set (r=0.78, p=0.038).

Whole-body glucose uptake (a) and liver glucose uptake (b) during SAL and IL/Hep studies. Uptake values are expressed per unit of body weight (M) and per unit of liver volume (HGU) respectively. * p<0.05 vs SAL

Plots for the estimation of HKi during the SAL study (a) and the IL/Hep study (b) showing a downward shift in the slope of the fitted line, but similar r values, in one study subject. a. Ki=0.0042; r=0.99. b. Ki=0.0019; r=0.99
The goodness of fit to the measured data of both graphical and modelling analysis was comparable in the SAL study (r value 0.98±0.01, SS 5.2±1.6 [kBq/ml]2) and the IL/Hep study (r value 0.96±0.02, SS 4.2±1.0 [kBq/ml]2; NS vs SAL).
Results from the three approaches for the detection of tracer outflow (outlined above) were consistent. Repeated linear fitting over progressive time intervals (approach 1) did not show any significant time-dependent decline in HKi or HGU, as shown in Figure 5, or any deterioration in the quality of the linear fit (r values). Values of kloss (approach 2; kloss=0 in 14 studies, kloss<0.001 in one SAL study) and k4 (approach 3; k4≤0.00002 in 14 studies, k4=0.001 in one SAL study) did not differ, either from each other or between the two studies (NS), and they approached the limit of detection, being almost equal to zero. Thus, recalculated, i.e. kloss-adjusted, HKi values using the modified versus the original linear model yielded overlapping results (δ=0 in each subject), demonstrating the lack of impact of kloss. Similarly, k4 was shown to be a negligible parameter, since k3 was much greater than k4, and since the k3:k4 ratio was 501±78 in the whole data set.

Linear fit of liver data over progressive time intervals in one study subject, showing no decline in hepatic glucose uptake (HGU) during the SAL study (open bars) or the IL/Hep study (closed bars). r>0.99 at all time points in both studies
As the estimates of HKi/HGU and k3 were obtained independently from graphical analysis and compartmental modelling respectively, it was possible to test their relationship. In all studies, both HKi and HGU were positively related to the phosphorylation rate constant k3 (Fig. 6).

Relationship between the phosphorylation parameter k3 and hepatic influx rate constant (HKi) (a) or glucose uptake (HGU) (b) during the SAL study (open squares) and the IL/Hep study (filled squares). a. r=0.75; p=0.0014. b. r=0.71; p=0.0029
Discussion
To the best of our knowledge, this is the first study to explore the influence of circulating NEFA on insulin-mediated liver glucose uptake in humans. Our data confirm that elevated NEFA impair whole-body glucose uptake, and demonstrate that this negative influence involves glucose disposal by the liver. Interestingly, the defect in HGU paralleled in magnitude the severity of the insulin resistance induced in peripheral tissues, potentially implicating common underlying mechanisms.
Because liver net balance measurements are impractical in humans due to the inaccessibility of the portal circulation, most human studies have been confined to the determination of glucose uptake across the entire splanchnic bed. Relevant to the present findings, splanchnic balance studies have been controversial [20, 21, 22], probably due to profound differences in study designs. These include the selected level of glycaemia, the oral versus intravenous route of glucose administration, and the direct versus indirect SGU measurement, the latter requiring assumptions on intestinal absorption of an oral glucose load. Thus, SGU was reported to be decreased in Type 2 diabetic patients [20] and either unchanged [21] or increased [22] in healthy subjects during IL/Hep infusion. In the current study, the liver was isolated from surrounding splanchnic tissues using PET imaging. Consistent with data from animal studies [17, 18], the present results emphasise the concept that extrapolating from all splanchnic organs to the liver might be misleading, especially during euglycaemia, when the contribution of HGU to total SGU is small [6, 23] and changes in HGU may remain undetected. Our findings are compatible with those of others [21] who failed to detect any change in extracellular glucose incorporation into glycogen during mild NEFA elevation and hyperglycaemic hyperinsulinaemia. Firstly, as stated by these authors, hyperglycaemia is a potent stimulator of HGU, and it might have obscured a defect in insulin action. Euglycaemia was maintained during the present study to eliminate this important confounder. Secondly, the uridine diphosphate glucose method does not account for glucose entering the glycolytic pathway, which is a major metabolic route of hepatic glucose-6-phosphate disposition during normoglycaemic hyperinsulinaemia [43, 44]. Instead, [18F]FDG PET measures the cumulative effect of all pathways of glucose disposal on glucose uptake by the targeted organ. Thirdly, a three-fold (present study) versus a less than two-fold [21] NEFA elevation may also make a difference. Because portal blood, i.e. the main fuel source to the liver, drains NEFA released from visceral fat, small changes in peripheral NEFA levels may cause negligible differences in portal NEFA levels. This study was planned to ensure a significant increment in the level of NEFA reaching the liver.
The main finding of the present study was that a moderate elevation of peripheral NEFA levels reduces insulin-stimulated HGU by approximately 25% in healthy men. This defect was of the same magnitude as that observed in dogs under similar conditions [17]. This result expands our limited knowledge of the in vivo regulation of HGU in humans, and by establishing an unequivocal cause–effect relationship, substantiates previous correlative findings between HGU and NEFA suppressibility in human and animal studies [16, 43]. Our finding supports the notion that the ability of insulin to stimulate HGU is partly mediated by its antilipolytic effect. However, it is worth noting that plasma insulin levels comparable to, or lower than, those of the present study increase HGU by 50–75% over fasting values [5, 6]. Therefore, at least half of the insulin action on HGU must be accounted for by mechanisms other than the lowering of plasma NEFA. An analogous conclusion was drawn in studies on dogs, where approximately half of the insulin-mediated increment in net HGU was attributed to the indirect action of the hormone [43].
The current study allows some insight into the molecular events mediating the observed effect of NEFA on HGU. In graphical analysis, the excellent linear fit to the measured data was indicative of the tracer entering an irreversible compartment during the SAL study, and to a significantly lesser extent, the IL/Hep study. Due to the study design and scan duration, we anticipated no tracer outflow as a confounding mechanism for the observed liver glucose retention. This expectation was confirmed by (i) the lack of a time-dependent HGU decline during progressive linear fitting intervals [41], (ii) the negligible magnitude of the tracer outflow parameters [40, 41], and (iii) the evidence that phosphorylation by far exceeded dephosphorylation [41]. Hence, selective tracing of glucose influx was attained, implicating this pathway in the NEFA-induced HGU changes reported here. Following this line of reasoning, the strong relationship observed between the phosphorylation rate constant and HGU is consistent with the suggested involvement of glucokinase [24, 25, 26] as a target of circulating NEFA. It has previously been shown that the exposure of liver cells to elevated long-chain fatty acids concentrations, either in vitro [24, 25] or in vivo in rats fed a high-fat diet [45], inhibited glucokinase activity. In rodents, the impairment in insulin-mediated glucokinase translocation and/or an allosteric inhibitory effect of NEFA on glucokinase was suggested to be an additional mechanism [26]. Interestingly, the design of the latter study was similar to that of the present one, including a 2-hour, three-fold NEFA elevation during euglycaemic hyperinsulinaemia.
A novel finding of the current study is that the decrement in whole-body glucose uptake was of the same magnitude as that in the liver. Moreover, the strong correlation between changes in HGU and changes in M speaks in favour of common targets. We have previously shown that the NEFA-induced decrease in whole-body glucose uptake reflects the decrease induced in skeletal muscle [15]. Because the regulation of glucose transport and phosphorylation differs profoundly between different organs, other metabolic steps are more likely to explain this association. This raises the possibility that the concept of a glucose/NEFA cycle, with its underlying mechanisms, may be extended to the liver. Previous evidence in this direction was provided by in vitro experiments, in which palmitate decreased the production of lactate, decreased the concentration of fructose-2,6-biphosphate, and decreased the flux through phosphofructokinase (the rate-limiting step of glycolysis) in hepatocytes [46]. It was proposed that increased citrate formation mediated, in part, this effect. Analogously, in dogs’ liver, uptake of circulating NEFA was reciprocally related to glycolytic and glucose oxidative fluxes, as reflected by a net output of lactate and CO2 from the liver [43]. Notably, these metabolic pathways are major sites of substrate competition in the heart and in skeletal muscle [14], and they are the preferred routes of glucose disposal in the liver under the conditions of the present study [43, 44]. Though the current technique does not allow direct dissection of intracellular events occurring downstream of glucose uptake and phosphorylation, it is intriguing that the percentage reduction in whole-body glucose disposal and HGU shown here corresponds approximately with the fraction of intracellular glucose entering glycolysis and/or glucose oxidation in respective tissues during euglycaemic hyperinsulinaemia [43, 47]. On the other hand, hyperinsulinaemia stimulates glycogen synthesis and inhibits glycogenolysis. An elevation of NEFA, at basal plasma insulin levels, did not affect glycogen synthesis in vitro [48], but it reduced glycogenolysis in vivo during euglycaemic euinsulinaemia [49]. No change in net glycogen synthesis was observed in dogs in the presence of concurrent inhibition of glycogenolysis by hyperglycaemia [18]. We could expect a similar effect in the present study due to hyperinsulinaemia. A smaller contribution of extracellular glucose than of newly synthesised glucose to the formation of glycogen was induced by NEFA elevation in dogs during hyperglycaemic euinsulinaemia [18], but not in humans during hyperglycaemic hyperinsulinaemia [21]. Given different study designs, it is difficult to extrapolate previous results to present studies; the possibility that glycogen synthesis contributed to the reduction in HGU observed here remains to be explored.
Overall, though the in vivo studies described here lack the numerosity and accuracy of measurement required to provide conclusive evidence on mechanisms underlying the observed effect of NEFA on HGU, our results are consistent with previous findings in suggesting that multiple targets may mediate the regulation of HGU by NEFA and insulin. Together with glucokinase, glycolytic/oxidative enzymes may be implicated during euglycaemia. The combined inhibition of glucose phosphorylation and of fructose-2,6-biphosphate formation in the above-mentioned palmitate experiment [46] lends support to the notion of this dual mechanism.
In summary, a three-fold elevation in circulating NEFA impaired insulin-mediated liver glucose uptake in healthy men. The observed relationships of HGU with the phosphorylation rate constant and with whole-body glucose uptake provide evidence in favour of multiple molecular mechanisms.
Abbreviations
- FDG:
-
2-fluoro-2-deoxyglucose
- HGU:
-
hepatic glucose uptake
- HKi:
-
influx rate constant
- M:
-
whole-body glucose uptake
- PET:
-
positron emission tomography
- ROI:
-
region of interest
- SGU:
-
splanchnic glucose uptake
- SS:
-
sum of squares
References
Cherrington AD (1999) Banting Lecture 1997. Control of glucose uptake and release by the liver in vivo. Diabetes 48:1198–1214
Radziuk J, Pye S (2001) Hepatic glucose uptake, gluconeogenesis and the regulation of glycogen synthesis. Diabetes Metab Res Rev 17:250–272
Barzilai N, Rossetti L (1993) Role of glucokinase and glucose-6-phosphatase in the acute and chronic regulation of hepatic glucose fluxes by insulin. J Biol Chem 268:25019–25025
Iynedjian P, Jotterand D, Nouspikel T, Asfari M, Pilot P (1989) Transcriptional induction of glucokinase gene by insulin in cultured liver cells and its repression by the glucagons-cAMP system. J Biol Chem 264:21824–21829
Edgerton DS, Cardin S, Emshwiller M et al. (2001) Small increases in insulin inhibit hepatic glucose production solely caused by an effect on glycogen metabolism. Diabetes 50:1872–1882
Iozzo P, Geisler F, Oikonen V et al. (2003) Insulin stimulates liver glucose uptake in humans: an 18F-FDG PET Study. J Nucl Med 44:682–689
Basu R, Basu A, Johnson C, Schwenk W, Rizza R (2003) Type 2 diabetes alters the insulin dose response curves for both stimulation of splanchnic glucose uptake (SGU) and suppression of endogenous glucose production (EGP). Diabetes 52 [Suppl 1]:A59
Arora P, Basu R, Zangeneh F, Schwenk W, Rizza R (2003) The ability of insulin to stimulate incorporation of extracellular glucose into hepatic glycogen is impaired in type 2 diabetes. Diabetes 52 [Suppl 1]:A59
Basu A, Basu R, Shah P et al. (2000) Effects of type 2 diabetes on the ability of insulin and glucose to regulate splanchnic and muscle glucose metabolism. Evidence for a defect in hepatic glucokinase activity. Diabetes 49:272–283
Basu A, Basu R, Shah P et al. (2001) Type 2 diabetes impairs splanchnic uptake of glucose but does not alter intestinal glucose absorption during enteral glucose feeding: additional evidence for a defect in hepatic glucokinase activity. Diabetes 50:1351–1362
Iozzo P, Hallsten K, Oikonen V et al. (2003) Insulin-mediated hepatic glucose uptake is impaired in type 2 diabetes: evidence for a relationship with glycaemic control. J Clin Endocrinol Metab 88:2055–2060
Caro JF, Triester S, Patel VK, Tapscott EB, Frazier NL, Dohm GL (1995) Liver glucokinase: decreased activity in patients with type II diabetes. Horm Metab Res 27:19–22
Boden G (1999) Free fatty acids, insulin resistance, and type 2 diabetes mellitus. Proc Assoc Am Physicians 111:241–248
Randle PJ (1998) Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev 14:263–283
Nuutila P, Koivisto VA, Knuuti J et al. (1992) Glucose-free fatty acid cycle operates in human heart and skeletal muscle in vivo. J Clin Invest 89:1767–1774
Iozzo P, Hallsten K, Oikonen V et al. (2003) Effects of metformin and rosiglitazone monotherapy on insulin-mediated hepatic glucose uptake and their relation to visceral fat in type 2 diabetes. Diabetes Care 26:2069–2074
Sindelar DK, Chu CA, Rohlie M, Neal DW, Swift LL, Cherrington AD (1997) The role of fatty acids in mediating the effects of peripheral insulin on hepatic glucose production in the conscious dog. Diabetes 46:187–196
Moore MC, Satake S, Lautz M et al. (2004) Nonesterified fatty acids and hepatic glucose metabolism in the conscious dog. Diabetes 53:32–40
Tonelli J, Kishore P, Stein D, Schubart U, Hawkins M (2003) Time-dependent effects of free fatty acids (FFA) on glucose effectiveness (GE) in type 2 diabetes mellitus (T2DM). Diabetes 52 [Suppl 1]:A59
Bajaj M, Pratipanawatr T, Berria R et al. (2002) Free fatty acids reduce splanchnic and peripheral glucose uptake in patients with type 2 diabetes. Diabetes 51:3043–3048
Shah P, Vella A, Basu A et al. (2002) Effects of free fatty acids and glycerol on splanchnic glucose metabolism and insulin extraction in nondiabetic humans. Diabetes 51:301–310
Bajaj M, Berria R, Pratipanawatr T et al. (2002) Free fatty acid-induced peripheral insulin resistance augments splanchnic glucose uptake in healthy humans. Am J Physiol Endocrinol Metab 283:E346–E352
DeFronzo RA (1987) Use of the splanchnic/hepatic balance technique in the study of glucose metabolism. Baillieres Clin Endocrinol Metab 1:837–862
Tippett PS, Neet KE (1982) Specific inhibition of glucokinase by long chain acyl coenzymes A below the critical micelle concentration. J Biol Chem 257:12839–12845
Tippett PS, Neet KE (1982) An allosteric model for the inhibition of glucokinase by long chain acyl coenzyme A. J Biol Chem 257:12846–12852
Lam TK, Werve G van de, Giacca A (2003) Free fatty acids increase basal hepatic glucose production and induce hepatic insulin resistance at different sites. Am J Physiol Endocrinol Metab 284:E281–E290
Massillon D, Barzilai N, Hawkins M, Prus-Wertheimer D, Rossetti L (1997) Induction of hepatic glucose-6-phosphatase gene expression by lipid infusion. Diabetes 46:153–157
Stingl H, Krssak M, Krebs M et al. (2001) Lipid-dependent control of hepatic glycogen stores in healthy humans. Diabetologia 44:48–54
Wiesenthal SR, Sandhu H, McCall RH et al. (1999) Free fatty acids impair hepatic insulin extraction in vivo. Diabetes 48:766–774
DeFronzo RA, Tobin JD, Andres R (1979) Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237:E214–E223
Hamacher K, Coenen HH, Stocklin G (1986) Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro-2-deoxy-D-glucose using aminopolyether supported nucleophilic substitution. J Nucl Med 27:235–238
van den Hoff J, Burchert W, Muller-Schauenburg W, Meyer GJ, Hundeshagen H (1993) Accurate local blood flow measurements with dynamic PET: fast determination of input function delay and dispersion by multilinear minimization. J Nucl Med 34:1770–1777
Patlak CS, Blasberg RG, Fenstermacher JD (1983) Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab 3:1–7
Munk OL, Bass L, Roelsgaard K, Bender D, Hansen SB, Keiding S (2001) Liver kinetics of glucose analogs measured in pigs by PET: importance of dual-input blood sampling. J Nucl Med 42:795–801
Keiding S, Hansen SB, Rasmussen HH et al. (1998) Detection of cholangiocarcinoma in primary sclerosing cholangitis by positron emission tomography. Hepatology 28:700–706
Keiding S, Munk OL, Schiott KM, Hansen SB (2000) Dynamic 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography of liver tumours without blood sampling. Eur J Nucl Med 27:407–412
Sokoloff L, Reivich M, Kennedy C et al. (1977) The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 28:897–916
Sweet IR, Peterson L, Kroll K, Goodner CJ, Berry M, Graham MM (1996) Effect of glucose on uptake of radiolabeled glucose, 2-DG, and 3-O-MG by the perfused rat liver. Am J Physiol 271:E384–E396
Bender D, Munk OL, Feng HQ, Keiding S (2001) Metabolites of (18)F-FDG and 3-O-(11)C-methylglucose in pig liver. J Nucl Med 42:1673–1678
Patlak CS, Blasberg RG (1985) Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J Cereb Blood Flow Metab 5:584–590
Lammertsma AA, Brooks DJ, Frackowiak RS et al. (1987) Measurement of glucose utilisation with [18F]2-fluoro-2-deoxy-D-glucose: a comparison of different analytical methods. J Cereb Blood Flow Metab 7:161–172
Choi Y, Hawkins RA, Huang SC et al. (1994) Evaluation of the effect of glucose ingestion and kinetic model configurations of FDG in the normal liver. J Nucl Med 35:818–823
Satake S, Moore MC, Igawa K et al. (2002) Direct and indirect effects of insulin on glucose uptake and storage by the liver. Diabetes 51:1663–1671
Halimi S, Assimacopoulos-Jeannet F, Terrettaz J, Jeanrenaud B (1987) Differential effect of steady-state hyperinsulinaemia and hyperglycaemia on hepatic glycogenolysis and glycolysis in rats. Diabetologia 30:268–272
Minassian C, Tarpin S, Mithieux G (1998) Role of glucose-6 phosphatase, glucokinase, and glucose-6 phosphate in liver insulin resistance and its correction by metformin. Biochem Pharmacol 55:1213–1219
Hue L, Maisin L, Rider MH (1988) Palmitate inhibits liver glycolysis. Involvement of fructose 2,6-bisphosphate in the glucose/fatty acid cycle. Biochem J 251:541–545
Ferrannini E, DeFronzo RA (1997) Insulin actions in vivo: glucose metabolism. In: Alberti KGGM, Zimmet P, DeFronzo RA (eds) International textbook of diabetes. Wiley, Chichester, pp 505–530
Gustafson LA, Neeft M, Reijngoud DJ et al. (2001) Fatty acid and amino acid modulation of glucose cycling in isolated rat hepatocytes. Biochem J 358:665–671
Chu CA, Sherck SM, Igawa K et al. (2002) Effects of free fatty acids on hepatic glycogenolysis and gluconeogenesis in conscious dogs. Am J Physiol Endocrinol Metab 282:E402–E411
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Iozzo, P., Lautamaki, R., Geisler, F. et al. Non-esterified fatty acids impair insulin-mediated glucose uptake and disposition in the liver. Diabetologia 47, 1149–1156 (2004). https://doi.org/10.1007/s00125-004-1443-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1443-2