
Abstract
The monoamine oxidase (MAO) enzymes metabolize neurotransmitter amines in the peripheral and central tissues, and inhibitors of these enzymes find application in the treatment of neuropsychiatric and neurodegenerative disorders. Based on reports that the neuronal nitric oxide synthase (nNOS) inhibitor, 7-nitroindazole, inhibits the MAO-B isoform, the present study investigated the MAO inhibition potencies of a synthetic series of fifteen C5- and C6-substituted indazole derivatives. While only one derivative (5c) was a submicromolar inhibitor of human MAO-A (IC50 = 0.745 µM), all compounds inhibited human MAO-B with submicromolar IC50 values. Substitution on C5 of indazole yielded particularly potent MAO-B inhibition with IC50 values that ranged from 0.0025–0.024 µM. Further investigation of a selected indazole derivative showed a competitive mode of MAO inhibition. To further explore the pharmacological properties of the indazole derivatives, they were also evaluated as potential inhibitors of porcine D-amino acid oxidase (DAAO). None of the synthetic compounds were noteworthy DAAO inhibitors, however, 1H-indazol-5-ol, a synthetic precursor, was found to be a good potency inhibitor with an IC50 value of 2.03 µM.

Similar content being viewed by others
Introduction
Various neurological disorders have been attributed to dysfunctional neurotransmission and include Parkinson’s disease, major depressive disorder and schizophrenia [1,2,3]. The observation that inhibition of enzymes such as monoamine oxidase (MAO; E.C. 1.4.3.4) and D-amino acid oxidase (DAAO; E.C. 1.4.3.3) increase monoaminergic and glutamatergic function, respectively, has led to an interest in the pharmacological potential of inhibitors of these enzymes in the palliative treatment of neuropsychiatric and neurodegenerative disorders [4, 5].
MAO is a mitochondrial bound flavoenzyme that is widely distributed throughout the body with high expression levels in both peripheral and nervous system tissues [6, 7]. These proteins are the primary degradative enzymes of monoamine neurotransmitters as well as various endogenous and exogenous amines [8]. MAO consists of two isozymes termed MAO-A and MAO-B. Even though these isozymes share 70% amino acid sequence homogeneity, MAO-A and MAO-B exhibit different substrate and inhibitor specificities [9, 10]. MAO-A metabolizes bulkier neurotransmitter amines such as serotonin, whereas MAO-B prefers smaller, non-hydroxylated amines as substrates (e.g., phenethylamine and benzylamine). Certain amines including dopamine, epinephrine and norepinephrine, tyramine and tryptamine are oxidized by both isozymes [5]. Primary amine substrates undergo MAO-catalyzed oxidative deamination to yield the corresponding aldehyde species with ammonia and hydrogen peroxide forming as by-products [11].
MAO regulates neurotransmitter levels and prevents certain extraneous molecules (e.g., dietary amines) from acting as false neurotransmitters, and metabolism facilitated by MAO is thus essential for the correct functioning at synaptic junctions [12]. Overactivity of MAO has been implicated in various psychiatric disorders. Elevated MAO-A density in different brain regions may be responsible for reducing monoamine levels (e.g., serotonin, norepinephrine and dopamine) during major depression and inhibitors of MAO-A have been used for the treatment of depression [13,14,15]. Similarly, MAO-B expression increases with age in the human brain, and MAO-B inhibitors have been used to treat age-related neurodegenerative disorders such as Parkinson’s disease [16, 17]. The rationale behind the use of MAO-B specific inhibitors is that these agents maintain synaptic dopamine levels by preventing its metabolism (or indirectly by elevating phenethylamine levels), thereby alleviating the motor symptoms of Parkinson’s disease [18, 19]. When combined with levodopa, MAO-B inhibitors may improve motor fluctuations and allow for a reduction of the levodopa dose [19]. Furthermore, MAO-mediated metabolism is a prominent source of hydrogen peroxide in the central nervous system and this by-product may contribute to neurodegeneration in Parkinson’s disease [20]. Under certain conditions, hydrogen peroxide might be converted to hydroxyl free radicals that cause oxidative stress and neuronal damage. This process has been linked with neurodegeneration observed in Parkinson’s disease and can potentially form part of the disease pathology [6, 21]. By attenuating hydrogen peroxide production, MAO inhibitors may thus possess neuroprotective effects and MAO-B inhibitors have been advocated as potential disease modifying agents [8, 22,23,24]. MAO-B inhibitors are a safe and well-tolerated approach in the symptomatic treatment of early or mild Parkinson’s disease and although these agents offer only modest symptomatic benefit, they reduce motor fluctuations caused by long-term use of levodopa, slow the rate of clinical decline and improve the efficacy of a lower levodopa dosage [19, 25, 26].
DAAO is a flavoenzyme that metabolizes D-amino acids such as D-alanine and D-serine [27, 28]. DAAO serves a regulatory function in the brain since it metabolizes the endogenous gliotransmitter, D-serine, which acts as a co-agonist that potentiates N-methyl-D-aspartate (NMDA) receptor-mediated neurotransmission by binding to its strychnine-insensitive glycine site [29, 30]. Hypofunction of the NMDA receptor has been implicated in the pathophysiology of schizophrenia, and D-serine and DAAO inhibitors have therefore been investigated for the treatment of schizophrenia [4, 31]. As adjuvant to antipsychotic drugs, D-serine has been shown to reduce the neurocognitive symptoms in schizophrenia [32, 33]. The poor blood-brain barrier permeability of D-serine, however, requires high doses to be administered which may lead to nephrotoxicity [34, 35]. DAAO inhibitors have been investigated as a potential treatment strategy for the positive and negative symptoms associated with schizophrenia. DAAO inhibitors would reduce the central metabolism of D-serine and thus enhance NMDA receptor-mediated neurotransmission [4, 36]. Most synthetic DAAO inhibitors are small molecules that contain the carboxylic acid group or isosteres thereof [31, 37, 38]. Several good potency DAAO inhibitors have been reported as exemplified by 3-hydroxycoumarin (1) and 3-hydroxyquinolin-2(1H)-one (2) (Fig. 1) [27, 31].

The structures of 3-hydroxycoumarin (1), 3-hydroxyquinolin-2(1H)-one (2) and 7-nitroindazole (3)
In previous studies, the neuronal nitric oxide synthase (nNOS) inhibitor, 7-nitroindazole (3), also demonstrated moderate MAO-B inhibition activity (Ki = 4 µM) [39, 40]. As with most small molecule inhibitors, 7-nitroindazole is expected to bind to the substrate cavity of the MAO-B enzyme, leaving the entrance cavity unoccupied [41,42,43]. In general, larger inhibitors that span both cavities are more potent due to the additional interactions formed between the inhibitor and the hydrophobic environment of the entrance cavity [43]. Appropriate substitution with a benzyloxy moiety has been reported to enhance the inhibitory efficacy of similar small molecules by enabling the compound to span both the entrance and substrate cavities of MAO-B [42, 44]. Based on these considerations, a series of C5- and C6-substituted indazole derivatives was synthesized in an attempt to improve the inhibition potency of 7-nitroindazole. The C5 and C6 positions were selected for substitution since literature showed that the MAO-B inhibition potency of another small molecule inhibitor, isatin, was significantly enhanced by substitution at the analogous positions [44, 45]. Substitution at these positions would direct the substituent into the entrance cavity. The series of indazole derivatives was evaluated as potential in vitro inhibitors of MAO-A, MAO-B and DAAO. Although there is no structural rationale for these compounds to act as DAAO inhibitors, the evaluation was used for comparison with the MAO inhibition potencies and for the generation of selectivity data.
Results and discussion
Chemistry
The indazole derivatives (4 and 5) were synthesized according to the method reported in literature. In the presence of K2CO3, commercially available 1H-indazol-5-ol (6) or 1H-indazol-6-ol (7) was dissolved in DMF and reacted with the appropriate alkyl bromides to produce the desired derivatives (Fig. 2). The yields of the reactions ranged from 3.1 to 22.8% (4a–i) and 10.2 to 18.4 (5a–f). The products were characterized by NMR and mass spectrometry (see supplementary material).

Synthetic route to C5- and C6-substituted indazoles 4a–i and 5a–f. Key: (i) DMF, K2CO3, rt, 24 h
Monoamine oxidase inhibition
The C5- and C6-substituted indazole derivatives synthesized in this study were evaluated as potential in vitro inhibitors of human MAO-A and MAO-B. The recombinant MAO enzymes were utilized to determine the inhibition properties of the compounds, while kynuramine served as a mixed MAO-A/B substrate. MAO metabolizes kynuramine to yield 4-hydroxyquinoline as final product. The production of 4-hydroxyquinoline was monitored by fluorescence spectrophotometry since the compound fluoresces in an alkaline environment. The enzyme reactions were thus carried out in the presence of a range of inhibitor concentrations (0.003–100 µM). After 20 min have elapsed, the reactions were terminated by the addition of sodium hydroxide and the formation of 4-hydroxyquinoline was quantitated. The enzyme catalytic activities were calculated and sigmoidal graphs of activity versus the logarithm of inhibitor concentration (log[I]) were constructed from triplicate experiments. These graphs were used to estimate IC50 values, which are reported in Table 1. Examples of sigmoidal graphs for the estimation of IC50 values are presented in Fig. 3.

A Sigmoidal graphs for the inhibition of MAO-A by 5c (filled circles), 5f (open circles) and toloxatone (triangles). B Sigmoidal graphs for the inhibition of MAO-B by 4d (filled circles), 5f (open circles) and safinamide (triangles)
The results of the inhibition studies showed that most of the indazole derivatives inhibited MAO-A with the most potent inhibition recorded for 5c (IC50 = 0.745 µM). This compound was the only submicromolar MAO-A inhibitor of the series. Among the test series, nine compounds exhibited IC50 < 10 µM and thus possessed potencies that were similar to the reference inhibitors isatin (IC50 = 8.29 µM) and toloxatone (IC50 = 1.67 µM). The following structure-activity relationships were noted for the inhibition of MAO-A: (a) The C6-substituted derivatives were more potent than the corresponding C5-substituted homologs (e.g., 4a vs. 5a; 4b vs. 5b; 4c vs. 5c; 4d vs. 5d; 4h vs. 5f). (b) For both the C5- and C6-substituted derivatives, the 4-chloro substituted compounds (4c and 5c) were the most potent MAO-A inhibitors while the methyl substituted compounds (4e, 4i, 5e) did not inhibit MAO-A at a maximal tested concentration of 100 µM. (c) It was interesting to note that neither 1H-indazol-5-ol (6) nor 1H-indazol-6-ol (7) inhibited MAO-A which demonstrated the requirement of a C5 or C6 substituent for inhibition.
The indazole derivatives were significantly more potent inhibitors of MAO-B compared to the MAO-A isoform, with all derivatives exhibiting IC50 < 1 µM. Based on their potency ranges, it was clear that the C5-substituted derivatives were more potent MAO-B inhibitors compared to the C6-substituted derivatives. For the C6-substituted derivatives, the IC50 values ranged from 0.118–0.979 µM while those of the C5-substituted derivatives ranged from 0.0025–0.024 µM, a difference of one to two orders of magnitude. The C5-substituted derivatives were therefore significantly more potent than the reference inhibitor safinamide (IC50 = 0.240 µM). The following structure-activity relationships were noted for the inhibition of MAO-B: (a) Among the C5-substituted derivatives, the benzyloxy substituted compound 4a (IC50 = 0.024 µM) was the weakest MAO-B inhibitor, which indicated that a substituent (F, Cl, Br, CH3, NO2) on the benzyloxy ring enhanced MAO-B inhibition. (b) For the C5-substituted derivatives, substitution on both the C4 and C3 positions of the benzyloxy ring (e.g., 4c vs. 4g; 4d vs. 4h; 4e vs. 4i) resulted in high potency inhibition, while for the C6-substituted derivatives (5a–f) substitution on C3 of the benzyloxy ring (5f) yielded the most potent inhibitor. In fact, 5f (IC50 = 0.118 µM) was approximately fivefold more potent than the C4-substituted homolog 5d (IC50 = 0.565 µM). (c) As for MAO-A, neither 1H-indazol-5-ol (6) nor 1H-indazol-6-ol (7) inhibited MAO-B which demonstrated the requirement of a C5 or C6 substituent for inhibition.
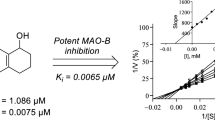
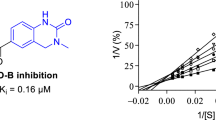
Since 7-nitroindazole is a reversible competitive inhibitor of MAO-B, it is likely that the indazole derivatives would also act as competitive inhibitors of the MAO enzymes. To further investigate the inhibition properties of the indazole derivative, sets of Lineweaver-Burk graphs were constructed for the inhibition of MAO-A and MAO-B by 5c. This compound represents a potent inhibitor of the respective MAO isoforms. To construct the Lineweaver-Burk graphs, MAO activities were measured in the absence and presence of the following set of inhibitor concentrations: ¼ × IC50, ½ × IC50, ¾ × IC50, 1 × IC50 and 1¼ × IC50. Each Lineweaver-Burk graph was prepared with substrate concentrations ranging from 15 to 250 μM. As shown in Fig. 4, the lines of the Lineweaver-Burk graphs intersected on the y-axis for both MAO-A and MAO-B, which was indicative of competitive inhibition. Replots of the slopes of the Lineweaver-Burk graphs versus inhibitor concentration also yielded linear lines from which the enzyme-inhibitor dissociation constants, Ki, were estimated (–Ki = x-axis intercept). Ki values of 0.40 and 0.16 µM were estimated for the inhibition of MAO-A and MAO-B, respectively, by 5c.

Lineweaver-Burk graphs for the inhibition of MAO-A (A) and MAO-B (B) by 5c. The insets are replots of the slopes of the Lineweaver-Burk plots versus inhibitor concentration, from which Ki values were estimated. For MAO-A the inhibitor concentration ranged from 0.186 to 0.931 µM, while for MAO-B, the inhibitor concentration ranged from 0.1 to 0.5 µM
Molecular docking
A molecular docking study was undertaken to investigate the potential binding modes and interactions of selected indazole derivatives (4a and 5a) in the active sites of MAO-A and MAO-B. For the docking study, the crystal structures of human MAO-A and MAO-B bound to harmine and safinamide, respectively, were used [42, 46]. The CDOCKER module of the Discovery Studio 3.1 suite of software was used and the protocol reported in literature was followed [47]. The predicted binding modes of 4a and 5a to MAO-A are presented in Fig. 5 and show that the inhibitors adopted virtually identical orientations in the active site, with the only difference being that the indazole ring is rotated through 180 ° between the two derivatives. As a result, 5a formed a hydrogen bond interaction with Asn-181 in addition to that observed with Tyr-444 for both derivatives. Other stabilizing interactions included pi interactions of the benzyloxy ring of the inhibitors with Phe-208 and both indazole rings with Tyr-407.

The predicted binding of 4a (A) and 5a (B) to the active site of human MAO-A. Hydrogen bonding is indicated by dashed lines. Residues Ile-199 and Tyr-326 in MAO-B, which correspond to Phe-208 and Ile-335 in MAO-A, respectively, are shown in green with the labels bracketed
The predicted binding modes of 4a and 5a to MAO-B are presented in Fig. 6. As for MAO-A, the inhibitors adopted similar binding orientations with the only difference being that the indazole ring was rotated through 180 °. A hydrogen bond interaction was observed between the inhibitors and Tyr-435. The MAO-B active site consists of two cavities, an entrance cavity which leads to a substrate cavity. For the inhibitors, the indazole ring was located in the substrate cavity while the benzyloxy ring extended into the entrance cavity. These compounds were thus cavity spanning inhibitors of MAO-B. Such compounds are reported to possess higher affinity for MAO-B due to the additional interactions provided by the entrance cavity compared to compounds that bind only to the substrate cavity [43]. Other interactions between the derivatives and MAO-B included a pi interaction between the phenyl of the indazole ring system and Tyr-326 and a pi-sulfur interaction between both rings of the indazole and Cys-172.

The predicted binding of 4a (A) and 5a (B) to the active site of human MAO-B. Hydrogen bonding is indicated by dashed lines. Residues Phe-208 and Ile-335 in MAO-A, which correspond to Ile-199 and Tyr-326 in MAO-B, respectively, are shown in green with the labels bracketed
D-Amino acid oxidase inhibition
DAAO from porcine kidney was used to investigate the inhibition properties of the indazole derivatives, while D-serine served as the substrate. The catalytic activity of DAAO was determined by measuring the production of H2O2 via continuous fluorescence spectrophotometry [37, 48]. The formation of H2O2 was measured in a peroxidase-coupled assay, where Amplex red is oxidized to produce the fluorescent compound, resorufin. DAAO and D-serine were incubated in the presence of various inhibitor concentrations (0.003–100 µM) while the fluorescence intensities were continuously monitored. Sigmoidal graphs of the rate of change in fluorescence vs. inhibitor concentration (log [I]) were constructed from triplicate experiments, and the IC50 values were calculated using these plots. The IC50 values are reported in Table 1 while Fig. 7 presents examples of sigmoidal graphs for the estimation of IC50 values.

Sigmoidal graphs for the inhibition of DAAO by 1H-indazol-5-ol (6) (filled circles) and 3-methylpyrazole-5-carboxylic acid (open circles)
The results showed that only five of the indazole derivatives exhibited weak DAAO inhibition with the most potent inhibition recorded for 4h (IC50 = 60.5 µM). Interestingly, all five of the compounds that showed inhibition were C5-substituted indazoles. However, 1H-indazol-5-ol (6) was found to be a good potency inhibitor with an IC50 value of 2.03 µM, whereas 1H-indazol-6-ol (7) did not inhibit the enzyme. The inhibition potency of 6 was thus similar to that of the reference inhibitor 3-methylpyrazole-5-carboxylic acid (IC50 = 2.04 µM).
There existed the possibility that a test compound may suppress the fluorescent signal produced by the peroxidase-coupled spectrofluorometric assay system, which may be incorrectly interpreted as DAAO inhibition. To investigate if this may have occurred for 1H-indazol-5-ol (6), the effect of the inhibitor on the fluorescence signal generated from hydrogen peroxide was measured. As shown in in Table 2, compound 6 decreased the fluorescent signal slightly at concentrations of 1 and 10 µM (92–96% residual signal), while significant suppression of the signal was observed at 100 µM (24% residual signal). Since the IC50 value for the inhibition of DAAO was well below 10 µM, it may be concluded that this compound was a true inhibitor and did not merely interfere with the assay system. For 3-methylpyrazole-5-carboxylic acid, only slight suppression of the fluorescence signal was observed at all tested concentrations.
The observation of the weak inhibition potencies of these compounds is consistent with reports that the binding site of DAAO prefers small and highly polar ligands [49]. Most of the known, good potency DAAO inhibitors are mono- or bicyclic compounds that consist of an acidic functionality (or isostere thereof) without large substituents or side-chains as exemplified by 3-hydroxycoumarin (1) and 3-hydroxyquinolin-2(1H)-one (2) [27, 31]. Despite the fact that the C5- and C6-substituted indazole derivatives are bicyclic, the bulky side-chains most likely prevented the compounds from binding and inhibiting DAAO.
Conclusion
It has been reported that 7-nitroindazole is a moderate MAO-B inhibitor. Since the inhibitory potencies of small molecule compounds can be enhanced with the addition of appropriate substituents, the present study synthesized and evaluated a series of C5 and C6-substituted indazole derivatives as in vitro inhibitors of MAO-A and MAO-B. The most potent inhibitors presented with IC50 values of 0.745 µM (5c) and 0.0025 µM (4d) for MAO-A and MAO-B, respectively. These compounds were more potent inhibitors than the MAO-A and MAO-B reference inhibitors, toloxatone (IC50 = 1.67 µM) and safinamide (IC50 = 0.240 µM), respectively. The results documented that the indazole derivatives were more potent MAO-B inhibitors than MAO-A inhibitors. Furthermore, the C5-substituted compounds displayed higher MAO-B inhibition potencies than the corresponding C6-substituted analogs. It may be concluded that the MAO-B inhibitors identified in this study could serve as lead compounds for the development of drugs for the treatment of Parkinson’s disease. However, none of the indazole derivatives exhibited good potency inhibition of DAAO. This might be attributed to the bulky side-chains resulting from substitution with the benzyloxy moiety that hindered the analogs from binding to the DAAO enzyme.
Experimental
Materials and instrumentation
Chemical reagents
The solvents and reagents used for the synthetic procedures were obtained from Sigma-Aldrich. Deuterated dimethylsulfoxide (DMSO-d6) used for nuclear magnetic resonance (NMR) spectroscopy was procured from Merck.
NMR
A Bruker Avance III 600 spectrometer was used to record proton (1H) and carbon (13C) NMR spectra at frequencies of 600 MHz and 150 MHz, respectively. The integration, multiplicity, and coupling constants (J), which are given in hertz (Hz), are included in the notation of the spectra. Chemical shifts (δ) are reported in parts per million (ppm) and were referenced to the residual solvent (DMSO-d6) signal at 2.5 ppm for 1H NMR and 39.5 ppm for 13C NMR. Spin multiplicities are denoted as s (singlet), d (doublet), dd (doublet of doublets), t (triplet) or m (multiplet).
Mass spectrometry
High resolution mass spectra (HRMS) were obtained with a Bruker micrOTOF-Q II mass spectrometer using atmospheric-pressure chemical ionization (APCI) in the positive mode.
TLC (thin layer chromatography)
The progression and completion of the chemical reactions were monitored by TLC. Silica gel 60 aluminum coated TLC sheets (containing UV254 fluorescent indicator) were used with a mobile phase consisting of ethyl acetate and hexane (3:2).
Biology
Enzymes, substrates and reference inhibitors used for the biological experiments were obtained from Sigma-Aldrich. A Varian Cary Eclipse fluorescence spectrophotometer (Agilent Technologies) and a SpectraMax iD3 multi-mode microplate reader (Molecular Devices) were used to record fluorescence measurements of all enzymatic reactions.
The synthesis of C5- (4a–i) and C6-substituted (5a–f) indazoles
1H-Indazol-5-ol or 1H-indazol-6-ol (4 mmol) was dissolved in anhydrous N,N-dimethylformamide (DMF, 10 mL) at room temperature, after which potassium carbonate (K2CO3, 8 mmol) was added. The appropriate alkyl bromide (5 mmol for 4a–f and 5a–f, 4.5 mmol for 4g–i) was subsequently added and the reaction was stirred at 0 °C for 2 h. The reaction was allowed to warm to room temperature and stirring was continued for 24 h. Upon completion, water (50 mL) was added and the resulting precipitate was collected by filtration and dried under vacuum. Compounds 5a–c were purified by recrystallisation from ethanol. Silica gel column chromatography (ethyl acetate:hexane, 4:1) was used to purify compounds 5d–f and 4a–i.
5‐(Benzyloxy)‐1H‐indazole (4a)
Yield: 10.0%; mp 175–177.4 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.94 (s, 1H), 7.95 (s, 1H), 7.50 – 7.44 (m, 3H), 7.43 – 7.37 (m, 2H), 7.36–7.31 (m, 1H), 7.29 – 7.24 (m, 1H), 7.08 (dd, J = 9.0, 2.3 Hz, 1H), 5.12 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 153.24, 137.77, 136.25, 133.29, 128.86, 128.20, 128.14, 123.51, 118.82, 111.53, 101.78, 70.09; APCI-HRMS m/z: calculated for C14H13N2O (MH+), 225.1022, found 225.1018.
5‐[(4‐Fluorophenyl)methoxy]‐1H‐indazole (4b)
Yield: 22.8%; mp 177.7-179.5 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.91 (s, 1H), 7.95 (s, 1H), 7.56 – 7.50 (m, 2H), 7.46 (d, J = 8.9 Hz, 1H), 7.29 – 7.25 (m, 1H), 7.25 – 7.19 (m, 2H), 7.08 (dd, J = 8.9, 2.3 Hz, 1H), 5.10 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 162.98, 161.37, 153.15, 136.32, 134.02, 134.00, 133.30, 130.36, 130.31, 123.51, 118.80, 115.72, 115.58, 111.52, 101.97, 69.47; APCI-HRMS m/z: calculated for C14H12FN2O (MH+), 243.0928, found 243.0924.
5‐[(4‐Chlorophenyl)methoxy]‐1H‐indazole (4c)
Yield: 19.0%; mp 179-181 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.91 (s, 1H), 7.94 (s, 1H), 7.51 (d, J = 8.4 Hz, 2H), 7.48 – 7.44 (m, 3H), 7.30 – 7.22 (m, 1H), 7.08 (dd, J = 8.9, 2.3 Hz, 1H), 5.13 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 153.05, 136.87, 136.34, 133.31, 132.75, 129.90, 128.85, 123.50, 118.76, 111.55, 102.03, 69.34; APCI-HRMS m/z: calculated for C14H12ClN2O (MH+), 259.0633, found 259.0626.
5‐[(4‐Bromophenyl)methoxy]‐1H‐indazole (4d)
Yield: 13.9%; mp 184–187 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.90 (s, 1H), 7.94 (s, 1H), 7.63 – 7.57 (m, 2H), 7.50 – 7.40 (m, 3H), 7.29 – 7.23 (m, 1H), 7.08 (dd, J = 8.9, 2.3 Hz, 1H), 5.11 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 153.04, 137.30, 136.33, 133.31, 131.78, 130.20, 123.50, 121.26, 118.75, 111.55, 102.05, 69.37; APCI-HRMS m/z: calculated for C14H12BrN2O (MH+), 303.0128, found 303.0142.
5‐[(4‐Methylphenyl)methoxy]‐1H‐indazole (4e)
Yield: 15.2%; mp 187-189.9 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.91 (s, 1H), 7.94 (s, 1H), 7.45 (d, J = 8.9 Hz, 1H), 7.36 (d, J = 7.9 Hz, 2H), 7.28 – 7.22 (m, 1H), 7.20 (d, J = 7.8 Hz, 2H), 7.06 (dd, J = 8.9, 2.3 Hz, 1H), 5.06 (s, 2H), 2.31 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 153.26, 137.40, 136.24, 134.73, 133.28, 129.40, 128.22, 123.52, 118.84, 111.48, 101.83, 70.03, 21.23; APCI-HRMS m/z: calculated for C15H15N2O (MH+), 239.1179, found 239.1168.
5‐[(4‐Nitrophenyl)methoxy]‐1H‐indazole (4f)
Yield: 5.8%; mp 191–193 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.95 (s, 1H), 8.27 (d, J = 8.8 Hz, 2H), 7.95 (s, 1H), 7.76 (d, J = 8.7 Hz, 2H), 7.48 (d, J = 9.0 Hz, 1H), 7.30 – 7.24 (m, 1H), 7.13 (dd, J = 9.0, 2.3 Hz, 1H), 5.31 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 152.81, 147.41, 145.81, 136.39, 133.34, 128.65, 124.05, 123.45, 118.68, 111.67, 102.09, 68.97; APCI-HRMS m/z: calculated for C14H12N3O3 (MH+), 270.0873, found 270.0863.
5‐[(3‐Chlorophenyl)methoxy]‐1H‐indazole (4g)
Yield: 3.1%; mp 163.5–165 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.93 (s, 1H), 7.95 (s, 1H), 7.56 – 7.53 (m, 1H), 7.47 (d, J = 9.0 Hz, 1H), 7.45 – 7.41 (m, 2H), 7.41–7.38 (m, 1H), 7.29 – 7.23 (m, 1H), 7.10 (dd, J = 9.0, 2.3 Hz, 1H), 5.15 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 152.98, 140.42, 136.33, 133.55, 133.32, 130.80, 128.10, 127.70, 126.59, 123.47, 118.75, 111.59, 101.96, 69.20; APCI-HRMS m/z: calculated for C14H12ClN2O (MH+), 259.0633, found 259.0637.
5‐[(3‐Bromophenyl)methoxy]‐1H‐indazole (4h)
Yield: 7.0%; mp 165–167 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.91 (s, 1H), 7.95 (s, 1H), 7.70 – 7.66 (m, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.51 – 7.44 (m, 2H), 7.37 (t, J = 7.8 Hz, 1H), 7.29 – 7.24 (m, 1H), 7.10 (dd, J = 9.0, 2.3 Hz, 1H), 5.14 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 153.00, 140.69, 136.36, 133.33, 131.08, 131.00, 130.57, 126.98, 123.49, 122.14, 118.74, 111.58, 102.03, 69.20; APCI-HRMS m/z: calculated for C14H12BrN2O (MH+), 303.0128, found 303.0142.
5‐[(3‐Methylphenyl)methoxy]‐1H‐indazole (4i)
Yield: 5.6%; mp 144–147 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.92 (s, 1H), 7.94 (s, 1H), 7.46 (d, J = 8.9 Hz, 1H), 7.31–7.28 (m, 2H), 7.28 – 7.24 (m, 2H), 7.14 (d, J = 7.1 Hz, 1H), 7.08 (dd, J = 8.9, 2.3 Hz, 1H), 5.07 (s, 2H), 2.33 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 153.30, 138.00, 137.69, 136.26, 133.30, 128.82, 128.76, 128.68, 125.22, 123.52, 118.81, 111.51, 101.76, 70.17, 21.47; APCI-HRMS m/z: calculated for C15H15N2O (MH+), 239.1179, found 239.1181.
6‐(Benzyloxy)‐1H‐indazole (5a)
Yield: 18.4%; mp 161–163 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.78 (s, 1H), 7.93 (s, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.52–7.46 (m, 2H), 7.41 (t, J = 7.9 Hz, 2H), 7.34 (t, J = 7.6 Hz, 1H), 7.05 – 6.99 (m, 1H), 6.83 (dd, J = 8.7, 2.1 Hz, 1H), 5.18 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 158.05, 141.45, 137.53, 133.82, 128.89, 128.24, 128.05, 121.72, 118.23, 113.12, 92.81, 69.94; APCI-HRMS m/z: calculated for C14H13N2O (MH+), 225.1022, found 225.1032.
6‐[(4‐Fluorophenyl)methoxy]‐1H‐indazole (5b)
Yield: 13.4%; mp 132–134 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.79 (s, 1H), 7.94 (s, 1H), 7.63 (d, J = 8.7 Hz, 1H), 7.57 – 7.50 (m, 2H), 7.23 (t, J = 8.9 Hz, 2H), 7.04 – 6.99 (m, 1H), 6.82 (dd, J = 8.8, 2.1 Hz, 1H), 5.16 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 163.03, 161.41, 157.94, 141.43, 133.83, 133.75, 133.73, 130.33, 130.27, 121.74, 118.27, 115.76, 115.62, 113.09, 92.84, 69.23; APCI-HRMS m/z: calculated for C14H12FN2O (MH+), 243.0928, found 243.0919.
6‐[(4‐Chlorophenyl)methoxy]‐1H‐indazole (5c)
Yield: 11.9%; mp 142.5–148 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.79 (s, 1H), 7.94 (s, 1H), 7.64 (d, J = 8.7 Hz, 1H), 7.54 – 7.49 (m, 2H), 7.48 – 7.44 (m, 2H), 7.03 – 6.98 (m, 1H), 6.83 (dd, J = 8.8, 2.2 Hz, 1H), 5.18 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 157.85, 141.41, 136.60, 133.83, 132.84, 129.84, 128.89, 121.77, 118.31, 113.06, 92.90, 69.11; APCI-HRMS m/z: calculated for C14H12ClN2O (MH+), 259.0633, found 259.0643.
6‐[(4‐Bromophenyl)methoxy]‐1H‐indazole (5d)
Yield: 10.6%; mp 167–169 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.83 (s, 1H), 7.94 (s, 1H), 7.64 (d, J = 8.8 Hz, 1H), 7.60 (d, J = 8.3 Hz, 2H), 7.45 (d, J = 8.4 Hz, 2H), 7.03 – 6.96 (m, 1H), 6.82 (dd, J = 8.7, 2.1 Hz, 1H), 5.16 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 157.80, 141.36, 137.01, 133.84, 131.83, 130.18, 121.79, 121.35, 118.26, 113.07, 92.79, 69.06; APCI-HRMS m/z: calculated for C14H12BrN2O (MH+), 303.0128, found 303.0115.
6‐[(4‐Methylphenyl)methoxy]‐1H‐indazole (5e)
Yield: 10.2%; mp 147–149 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.83 (s, 1H), 7.94 (s, 1H), 7.62 (d, J = 8.8 Hz, 1H), 7.37 (d, J = 7.9 Hz, 2H), 7.21 (d, J = 7.8 Hz, 2H), 7.02 – 6.97 (m, 1H), 6.80 (dd, J = 8.7, 2.1 Hz, 1H), 5.12 (s, 2H), 2.31 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 158.03, 141.41, 137.50, 134.43, 133.81, 129.45, 128.19, 121.71, 118.12, 113.19, 92.65, 69.75, 21.24; APCI-HRMS m/z: calculated for C15H15N2O (MH+), 239.1179, found 239.1169.
6‐[(3‐Bromophenyl)methoxy]‐1H‐indazole (5f)
Yield: 12.4%; mp 146.7–149 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.82 (s, 1H), 7.94 (s, 1H), 7.71 – 7.68 (m, 1H), 7.64 (d, J = 8.8 Hz, 1H), 7.57 – 7.47 (m, 2H), 7.38 (t, J = 7.8 Hz, 1H), 7.03 – 6.98 (m, 1H), 6.84 (dd, J = 8.7, 2.1 Hz, 1H), 5.20 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 157.77, 141.37, 140.40, 133.84, 131.13, 131.08, 130.56, 126.94, 122.16, 121.82, 118.31, 113.06, 92.83, 68.91; APCI-HRMS m/z: calculated for C14H12BrN2O (MH+), 303.0128, found 303.0129.
MAO activity measurements
The activities of MAO-A and MAO-B were measured by following the procedure as reported in literature [47, 50]. For this purpose, kynuramine was used as a mixed substrate and commercially available recombinant human MAO-A and MAO-B served as enzyme sources. Kynuramine is oxidized by MAO to produce 4-hydroxyquinoline as final product. Fluorescence spectrophotometry was used to measure 4-hydroxyquinoline following the alkalinization of the reactions at the end-point.
DAAO activity measurements
To measure the activity of DAAO, a previously reported protocol was followed [37, 48, 51]. D-Serine served as the substrate and porcine kidney DAAO was used as enzyme source. H2O2, a by-product of the catalytic cycle of DAAO, was measured in a peroxidase-coupled assay system using the reagent, Amplex red. In the presence of H2O2, horseradish peroxidase catalyzes the oxidation of Amplex red to yield the fluorescent compound, resorufin. Fluorescence spectrophotometry was used to continuously measure resorufin formation.
To investigate the possibility that a test inhibitor may suppress the fluorescence signal produced in the peroxidase-coupled assay system, the test inhibitor at 1, 10 and 100 µM was incubated with horse radish peroxidase, Amplex red and hydrogen peroxide and the fluorescence intensities were measured. These data were compared to control experiments conducted in the absence of inhibitor. The protocol for this experiment has been reported [51].
Molecular docking
Molecular docking was performed according to the protocol previously described [47]. The Discovery Studio 3.1 suite was used and X-ray crystal structures of MAO-A (PDB code: 2Z5X) and MAO-B (PDB code: 2V5Z) bound to harmine and safinamide, respectively, were selected as the protein models [42, 46]. Illustrations were created with the PyMOL molecular graphics system [52].
Abbreviations
- DAAO:
-
D-amino acid oxidase
- DMF:
-
N,N-dimethylformamide
- DMSO:
-
dimethyl sulfoxide
- HRMS:
-
high resolution mass spectrometry
- MAO:
-
monoamine oxidase
- NMDA:
-
N-methyl-D-aspartate
- NMR:
-
nuclear magnetic resonance
- nNOS:
-
neuronal nitric oxide synthase
- SD:
-
standard deviation
- TLC:
-
thin layer chromatography
References
Barone P. Neurotransmission in Parkinson’s disease: beyond dopamine. Eur J Neurol. 2010;17:364–76. https://doi.org/10.1111/j.1468-1331.2009.02900.x
Millan MJ. N-Methyl-D-aspartate receptors as a target for improved antipsychotic agents: novel insights and clinical perspectives. Psychopharmacology (Berl). 2005;179:30–53. https://doi.org/10.1007/s00213-005-2199-1
Werner FM, Covenas R. Classical neurotransmitters and neuropeptides involved in major depression: a review. Int J Neurosci. 2010;120:455–70. https://doi.org/10.3109/00207454.2010.483651
Lane HY, Lin CH, Green MF, Hellemann G, Huang CC, Chen PW, et al. Add-on treatment of benzoate for schizophrenia: a randomized, double-blind, placebo-controlled trial of D-amino acid oxidase inhibitor. JAMA Psychiatry. 2013;70:1267–75. https://doi.org/10.1001/jamapsychiatry.2013.2159
Youdim MB, Edmondson D, Tipton KF. The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci. 2006;7:295–309. https://doi.org/10.1038/nrn1883
Fowler JS, MacGregor RR, Wolf AP, Arnett CD, Dewey SL, Schlyer D, et al. Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science. 1987;235:481–5. https://doi.org/10.1126/science.3099392
Rodriguez MJ, Saura J, Billett EE, Finch CC, Mahy N. Cellular localization of monoamine oxidase A and B in human tissues outside of the central nervous system. Cell Tissue Res. 2001;304:215–20. https://doi.org/10.1007/s004410100361
Youdim MB, Bakhle YS. Monoamine oxidase: isoforms and inhibitors in Parkinson’s disease and depressive illness. Br J Pharmacol. 2006;147:S287–96. https://doi.org/10.1038/sj.bjp.0706464
Bach AW, Lan NC, Johnson DL, Abell CW, Bembenek ME, Kwan SW, et al. cDNA cloning of human liver monoamine oxidase A and B: molecular basis of differences in enzymatic properties. Proc Natl Acad Sci USA. 1988;85:4934–8. https://doi.org/10.1073/pnas.85.13.4934
Shih JC. Monoamine oxidase isoenzymes: genes, functions and targets for behavior and cancer therapy. J Neural Transm (Vienna). 2018;125:1553–66. https://doi.org/10.1007/s00702-018-1927-8
Edmondson DE. Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: biological implications. Curr Pharm Des. 2014;20:155–60. https://doi.org/10.2174/13816128113190990406
Bortolato M, Chen K, Shih JC. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv Drug Deliv Rev. 2008;60:1527–33. https://doi.org/10.1016/j.addr.2008.06.002
Meyer JH, Ginovart N, Boovariwala A, Sagrati S, Hussey D, Garcia A, et al. Elevated monoamine oxidase a levels in the brain: an explanation for the monoamine imbalance of major depression. Arch Gen Psychiatry. 2006;63:1209–16. https://doi.org/10.1001/archpsyc.63.11.1209
Shulman KI, Herrmann N, Walker SE. Current place of monoamine oxidase inhibitors in the treatment of depression. CNS Drugs. 2013;27:789–97. https://doi.org/10.1007/s40263-013-0097-3
Suchting R, Tirumalajaru V, Gareeb R, Bockmann T, de Dios C, Aickareth J, et al. Revisiting monoamine oxidase inhibitors for the treatment of depressive disorders: A systematic review and network meta-analysis. J Affect Disord. 2021;282:1153–60. https://doi.org/10.1016/j.jad.2021.01.021
Fowler CJ, Wiberg A, Oreland L, Marcusson J, Winblad B. The effect of age on the activity and molecular properties of human brain monoamine oxidase. J Neural Transm. 1980;49:1–20. https://doi.org/10.1007/BF01249185
Tong J, Meyer JH, Furukawa Y, Boileau I, Chang LJ, Wilson AA, et al. Distribution of monoamine oxidase proteins in human brain: implications for brain imaging studies. J Cereb Blood Flow Metab. 2013;33:863–71. https://doi.org/10.1038/jcbfm.2013.19
Berry MD, Scarr E, Zhu MY, Paterson IA, Juorio AV. The effects of administration of monoamine oxidase-B inhibitors on rat striatal neurone responses to dopamine. Br J Pharmacol. 1994;113:1159–66. https://doi.org/10.1111/j.1476-5381.1994.tb17119.x
Robakis D, Fahn S. Defining the role of the monoamine oxidase-B inhibitors for Parkinson’s disease. CNS Drugs. 2015;29:433–41. https://doi.org/10.1007/s40263-015-0249-8
Pizzinat N, Copin N, Vindis C, Parini A, Cambon C. Reactive oxygen species production by monoamine oxidases in intact cells. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:428–31. https://doi.org/10.1007/pl00005371
Cohen G. Monoamine oxidase and oxidative stress at dopaminergic synapses. J Neural Transm Suppl. 1990;32:229–38. https://doi.org/10.1007/978-3-7091-9113-2_33
Fernandez HH, Chen JJ. Monoamine oxidase-B inhibition in the treatment of Parkinson’s disease. Pharmacotherapy. 2007;27:174S–85S. https://doi.org/10.1592/phco.27.12part2.174S
Finberg JP, Rabey JM. Inhibitors of MAO-A and MAO-B in psychiatry and neurology. Front Pharmacol. 2016;7:340. https://doi.org/10.3389/fphar.2016.00340
Kumar B, Gupta VP, Kumar V. A perspective on monoamine oxidase enzyme as drug target: challenges and opportunities. Curr Drug Targets. 2017;18:87–97. https://doi.org/10.2174/1389450117666151209123402
Stowe R, Ives N, Clarke CE, Handley K, Furmston A, Deane K, et al. Meta-analysis of the comparative efficacy and safety of adjuvant treatment to levodopa in later Parkinson’s disease. Mov Disord. 2011;26:587–98. https://doi.org/10.1002/mds.23517
Tan YY, Jenner P, Chen SD. Monoamine oxidase-B inhibitors for the treatment of Parkinson’s disease: past, present, and future. J Parkinsons Dis. 2022;12:477–93. https://doi.org/10.3233/JPD-212976
Katane M, Osaka N, Matsuda S, Maeda K, Kawata T, Saitoh Y, et al. Identification of novel D-amino acid oxidase inhibitors by in silico screening and their functional characterization in vitro. J Med Chem. 2013;56:1894–907. https://doi.org/10.1021/jm3017865
Pollegioni L, Sacchi S, Murtas G. Human D-amino acid oxidase: structure, function, and regulation. Front Mol Biosci. 2018;5:107. https://doi.org/10.3389/fmolb.2018.00107
Mothet JP, Parent AT, Wolosker H, Brady RO Jr, Linden DJ, Ferris CD, et al. D-Serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci USA. 2000;97:4926–31. https://doi.org/10.1073/pnas.97.9.4926
Wolosker H. NMDA receptor regulation by D-serine: new findings and perspectives. Mol Neurobiol. 2007;36:152–64. https://doi.org/10.1007/s12035-007-0038-6
Duplantier AJ, Becker SL, Bohanon MJ, Borzilleri KA, Chrunyk BA, Downs JT, et al. Discovery, SAR, and pharmacokinetics of a novel 3-hydroxyquinolin-2(1H)-one series of potent D-amino acid oxidase (DAAO) inhibitors. J Med Chem. 2009;52:3576–85. https://doi.org/10.1021/jm900128w
Heresco-Levy U, Javitt DC, Ebstein R, Vass A, Lichtenberg P, Bar G, et al. D-Serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol Psychiatry. 2005;57:577–85. https://doi.org/10.1016/j.biopsych.2004.12.037
Kantrowitz JT, Malhotra AK, Cornblatt B, Silipo G, Balla A, Suckow RF, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res. 2010;121:125–30. https://doi.org/10.1016/j.schres.2010.05.012
Maekawa M, Okamura T, Kasai N, Hori Y, Summer KH, Konno R. D-amino-acid oxidase is involved in D-serine-induced nephrotoxicity. Chem Res Toxicol. 2005;18:1678–82. https://doi.org/10.1021/tx0500326
Nishikawa T. Analysis of free D-serine in mammals and its biological relevance. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:3169–83. https://doi.org/10.1016/j.jchromb.2011.08.030
Ferraris D, Duvall B, Ko YS, Thomas AG, Rojas C, Majer P, et al. Synthesis and biological evaluation of D-amino acid oxidase inhibitors. J Med Chem. 2008;51:3357–9. https://doi.org/10.1021/jm800200u
Hondo T, Warizaya M, Niimi T, Namatame I, Yamaguchi T, Nakanishi K, et al. 4-Hydroxypyridazin-3(2H)-one derivatives as novel D-amino acid oxidase inhibitors. J Med Chem. 2013;56:3582–92. https://doi.org/10.1021/jm400095b
Sparey T, Abeywickrema P, Almond S, Brandon N, Byrne N, Campbell A, et al. The discovery of fused pyrrole carboxylic acids as novel, potent D-amino acid oxidase (DAO) inhibitors. Bioorg Med Chem Lett. 2008;18:3386–91. https://doi.org/10.1016/j.bmcl.2008.04.020
Castagnoli K, Palmer S, Anderson A, Bueters T, Castagnoli N Jr. The neuronal nitric oxide synthase inhibitor 7-nitroindazole also inhibits the monoamine oxidase-B-catalyzed oxidation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Chem Res Toxicol. 1997;10:364–8. https://doi.org/10.1021/tx970001d
Di Monte DA, Royland JE, Anderson A, Castagnoli K, Castagnoli N Jr, Langston JW. Inhibition of monoamine oxidase contributes to the protective effect of 7-nitroindazole against MPTP neurotoxicity. J Neurochem. 1997;69:1771–3. https://doi.org/10.1046/j.1471-4159.1997.69041771.x
Binda C, Li M, Hubalek F, Restelli N, Edmondson DE, Mattevi A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc Natl Acad Sci USA. 2003;100:9750–5. https://doi.org/10.1073/pnas.1633804100
Binda C, Wang J, Pisani L, Caccia C, Carotti A, Salvati P, et al. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: safinamide and coumarin analogs. J Med Chem. 2007;50:5848–52. https://doi.org/10.1021/jm070677y
Hubalek F, Binda C, Khalil A, Li M, Mattevi A, Castagnoli N, et al. Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors. J Biol Chem. 2005;280:15761–6. https://doi.org/10.1074/jbc.M500949200
Manley-King CI, Bergh JJ, Petzer JP. Inhibition of monoamine oxidase by selected C5- and C6-substituted isatin analogues. Bioorg Med Chem. 2011;19:261–74. https://doi.org/10.1016/j.bmc.2010.11.028
Van der Walt EM, Milczek EM, Malan SF, Edmondson DE, Castagnoli N Jr, Bergh JJ, et al. Inhibition of monoamine oxidase by (E)-styrylisatin analogues. Bioorg Med Chem Lett. 2009;19:2509–13. https://doi.org/10.1016/j.bmcl.2009.03.030
Son SY, Ma J, Kondou Y, Yoshimura M, Yamashita E, Tsukihara T. Structure of human monoamine oxidase A at 2.2-A resolution: the control of opening the entry for substrates/inhibitors. Proc Natl Acad Sci USA. 2008;105:5739–44. https://doi.org/10.1073/pnas.0710626105
Mostert S, Petzer A, Petzer JP. Indanones as high-potency reversible inhibitors of monoamine oxidase. ChemMedChem. 2015;10:862–73. https://doi.org/10.1002/cmdc.201500059
Zhou M, Panchuk-Voloshina N. A one-step fluorometric method for the continuous measurement of monoamine oxidase activity. Anal Biochem. 1997;253:169–74. https://doi.org/10.1006/abio.1997.2392
Molla G. Competitive inhibitors unveil structure/function relationships in human D-amino acid oxidase. Front Mol Biosci. 2017;4:80. https://doi.org/10.3389/fmolb.2017.00080
Weissbach H, Smith TE, Daly JW, Witkop B, Udenfriend S. A rapid spectrophotometric assay of mono-amine oxidase based on the rate of disappearance of kynuramine. J Biol Chem. 1960;235:1160–3
Bester E, Petzer A, Petzer JP. Coumarin derivatives as inhibitors of d-amino acid oxidase and monoamine oxidase. Bioorg Chem. 2022;123:105791. https://doi.org/10.1016/j.bioorg.2022.105791
DeLano WL. The PyMOL molecular graphics system. San Carlos, USA: DeLano Scientific; 2002
Acknowledgements
This work was financially supported by the National Research Foundation of South Africa [Grant specific unique reference numbers (UID) 137997 and 132168]. The Grantholders acknowledge that opinions, findings and conclusions or recommendations expressed in any publication generated by the NRF supported research are that of the authors, and that the NRF accepts no liability whatsoever in this regard.
Funding
Open access funding provided by North-West University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stear, C., Petzer, A., Crous, C. et al. Indazole derivatives as novel inhibitors of monoamine oxidase and D-amino acid oxidase. Med Chem Res 33, 164–176 (2024). https://doi.org/10.1007/s00044-023-03176-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-023-03176-x