
Abstract
The limited endogenous regenerative capacity of the human heart renders cardiovascular diseases a major health threat, thus motivating intense research on in vitro heart cell generation and cell replacement therapies. However, so far, in vitro-generated cardiomyocytes share a rather fetal phenotype, limiting their utility for drug testing and cell-based heart repair. Various strategies to foster cellular maturation provide some success, but fully matured cardiomyocytes are still to be achieved. Today, several hormones are recognized for their effects on cardiomyocyte proliferation, differentiation, and function. Here, we will discuss how the endocrine system impacts cardiomyocyte maturation. After detailing which features characterize a mature phenotype, we will contemplate hormones most promising to induce such a phenotype, the routes of their action, and experimental evidence for their significance in this process. Due to their pleiotropic effects, hormones might be not only valuable to improve in vitro heart cell generation but also beneficial for in vivo heart regeneration. Accordingly, we will also contemplate how the presented hormones might be exploited for hormone-based regenerative therapies.
Graphical abstract

Similar content being viewed by others
Introduction
The endocrine system fulfills messenger functions via the circulatory system and mediates partially long-lasting regulatory cues. As such, it complements the rapid transmission of transient signals via the nervous system. A plethora of hormones produced by endocrine glands and organs governs organismal development, crucial functions such as metabolism, homeostasis, and reproduction as well as adaptations to environmental pressures. Major parts of the endocrine system comprise the hypothalamus, pituitary gland, pineal body, thyroid and parathyroid gland, thymus, adrenal gland, pancreas, ovary, and testis, while recently further organs are recognized for their production of hormones, including the heart [205]. The heart owes its attribution as an endocrine organ primarily due to the secretion of the polypeptide hormones atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) by atrial cardiomyocytes. However, a broad range of heart-derived hormones was discovered [205, 325], demonstrating systemic and local effects as will be briefly described in this review.
Effects of the endocrine system on heart development [100, 136] and function [25, 238] are well known, and indications for a significant role of hormones in cardiovascular diseases consolidate. For example, it has been known for decades that women are at lower risk of developing ischemic heart disease, which is in part attributed to protective effects of estrogens [29, 143]. However, estrogen cannot fully explain this sex dimorphism as a meta-study reports that estrogen does not provide a clear beneficial effect on heart disease incidence in healthy men [293]. This is just one example of how hormonal effects and underlying mechanisms are still not fully understood, which holds especially true on the cellular level. Nevertheless, we have started understanding the diverse effects of selected hormones on cardiomyocyte remodeling under physiological and pathological conditions [55].
Globally, ischemic heart disease poses a leading cause of death [148] as affected heart muscle tissue suffers from ischemia–reperfusion injury [104] and is mostly irreversibly lost due to the limited regenerative capacity of adult cardiomyocytes. Intriguingly, a study across 42 species with varying degrees of regenerative capacities reveals that certain morphological requirements to regenerate heart tissue, such as cardiomyocyte diploidy, correlate with rates of metabolism, body temperature, and serum thyroid levels [119]. Moreover, the authors demonstrate that diminishing thyroid hormone signaling could retain cardiac regenerative potential in mice, while promoting high thyroid levels inhibits tissue regeneration in zebrafish [119]. As endothermy is accompanied by increased thyroid hormone levels, Hirose et al. suggest that the loss of regenerative capacity in adult mammals is an evolutionary trade-off for the acquisition of endothermy.
Irrespective of the origin, the lack of endogenous regenerative capacity due to the very limited turnover rate of cardiomyocytes in humans [20] is an issue that motivated much research on cell replacement strategies for damaged heart tissue. Today, induced pluripotent stem cells (iPSCs) are frequently used to derive cardiomyocytes as model systems and for therapeutic approaches [134]. Cardiomyocytes can be generated from iPSCs by a number of different protocols, usually relying on stage-specific activation and inhibition of different signaling pathways, such as WNT and NOTCH signaling [34]. However, so far, the in vitro-generated cardiomyocytes share a rather fetal phenotype regardless of the utilized approach to attain them [19, 34, 65, 292]. Recently, single-cell-based approaches and maturation metrics have aided a more holistic and quantitative evaluation of cellular maturity, confirming that the majority of human pluripotent stem cell-derived cardiomyocytes (hPSC-CMs) fail to resemble adult cardiomyocytes [198].
Hence, different strategies have been developed to foster the maturation of cardiomyocytes, as comprehensively summarized by others [107, 296]. Long-term culture [140, 166], 3D systems, and extracellular matrices [6], as well as electromechanical stimulation [204, 235, 242], provide limited success. However, complete maturation could not be achieved, which may be attributed to a lack of environmental signals. In line with this, the implantation of immature cardiomyocytes into the hearts of non-human primates results in progressive maturation [49, 112]. As mentioned above, in vivo, many processes are orchestrated by the endocrine system that modulates the cellular environment via the circulatory system. Here, we will primarily focus on the endocrine control of cardiomyocyte maturation. After detailing which features characterize a mature phenotype, we will contemplate hormones most promising to induce such a phenotype, the routes of their action, and experimental evidence for their significance in this process. As some of the presented hormones are also implicated in cardioprotection and regeneration, we will additionally explore these different aspects of their action and how they could be exploited for hormone-based regenerative therapies.
Hallmarks of mature cardiomyocytes
After birth, environmental conditions and demands on the heart are dramatically changing. Cardiac cells are exposed to higher oxygen levels and have to cope with increased blood pressure and mechanical load. Consequently, extensive modifications concerning the cellular composition but also individual cell type features commence to adapt to these new demands [123, 264]. The vast majority of cardiomyocytes exit the cell cycle (G0 phase), initiating essential metabolic, structural, and electrophysical maturation steps but also inevitably losing regenerative capacity in the process.
In mice, the last round of the cell cycle includes karyokinesis but no cytokinesis, which results in around 90% of mature cardiomyocytes having two diploid nuclei [168, 266]. In humans, only around 25% of mature cardiomyocytes are binucleated. However, most nuclei are polyploid due to DNA endoreplication without karyokinesis [30, 207]. Accordingly, the remaining mononuclear diploid cardiomyocytes that are suggested to drive heart regeneration [22, 159, 225] are a minor subpopulation both in murine and human adult hearts. This proliferation-to-hypertrophy transition in postnatal hearts is an indicator of the underlying maturation processes on the cellular level that will be covered here. Notably, pharmacologically shifting human iPSCs to the G0 phase by transient inhibition of the mammalian target of rapamycin (mTOR) signaling pathway results in enhanced cardiomyocyte maturation [80], stressing the causality between cell cycle exit and maturation.
Metabolic maturation
Postnatally, changes in cardiac metabolism are required to meet the increased energy demand for maintaining contractile function. Upon birth, the increase in levels of both oxygen and free fatty acids originating from the lipid content in maternal milk allows for a switch from glycolysis to the more efficient oxidative phosphorylation for ATP production [179]. Importantly, this “metabolic switch” is not merely an effect of cardiomyocyte maturation but a key driver of this process. Indeed, several groups found that fatty acid supplementation could promote cell cycle arrest and maturation of hPSC-CMs in vitro [53, 121, 190, 320], while high glucose inhibits this process [200]. Moreover, a recent in vivo study in mice demonstrates that the essential fatty acid γ-linolenic acid, which has been found to be enriched in maternal milk, induces a regulatory mechanism triggering the metabolic switch. Conversely, interference of the downstream retinoid X receptors results in perinatal cardiac dysfunction [219].
The transition of the metabolic phenotype toward mitochondrial oxidative metabolism is mainly coordinated by ligand-dependent nuclear receptor pathways. The three most relevant pathways for this process include the hypoxia-inducible factor (HIF-1α/2α) pathway [188], the peroxisome proliferator-activated receptor (PPAR) γ coactivator 1α (PGC1α)/PPARα pathway [165, 271], and the PGC1α/PPARβ/δ pathway [33, 271]. Crosstalk between HIF-1α/2α and metabolic sensors, such as the AMP-activated kinase (AMPK) and mTOR, modulates the response of cardiomyocytes to the altered substrates and can even impact mitochondrial dynamics and morphology [81]. Most prominently, HIF-1α activity enhances lactate dehydrogenase A expression and promotes glycolysis [256, 285]. Conversely, inhibition of this axis supports the switch from glycolysis to oxidative phosphorylation and has been shown to improve metabolic maturation in hPSC-CMs [122]. In contrast, the PGC1α/PPAR pathways promote metabolic maturation via YAP1 and SF3B2, as recently demonstrated at single-cell level [199]. In hPSC-CMs, PPARδ activation fosters fatty acid oxidation and has been demonstrated to enhance metabolic as well as structural maturation [313].
Moreover, ligand-independent nuclear receptor pathways, such as the estrogen-related receptor (ERR) pathway, are found to be vital for the transition to oxidative metabolism [4, 246]. ERRγ expression affects transcription factors, such as PPARγ, PPARδ, Foxo1, and Gata4, and regulates a nuclear-encoded mitochondrial gene network, thereby impacting mitochondrial oxidative phosphorylation and ion transport [4, 246].
The metabolic maturation of cardiomyocytes is also accompanied by isoform switching of some implicated enzymes. For example, hexokinase (HK), which catalyzes the initial step in glycolysis, switches from HK1 to the less active form HK2 [37], while cytochrome c oxidase subunit 8 (COX8), the last enzyme in the mitochondrial electron transport chain, switches from COX8A to COX8B [65]. However, the functional relevance of this switch for cardiomyocyte maturation remains unclear. In adipose tissue, COX8B expression is associated with thermogenic differentiation [75, 82], and thus, the isoform switch might just reflect an unrelated effect due to the progression of endothermy after birth. Another study in glomus cells of the carotid body hypothesizes that atypical cytochrome oxidase subunits facilitate acute O2 sensing [195]. A link between this isoform switch and the altered substrate utilization upon cardiomyocyte maturation is not yet established.
Finally, the sites of oxidative phosphorylation, mitochondria, undergo extensive modifications in number, size, and structure. These adaptations for higher ATP production rates are linked to further structural changes and will be outlined in the following.
Structural maturation
The high levels of ATP production in mature cardiomyocytes are reflected by the large mitochondrial content taking up around 30% of the volume in the myocardium of adult rodents and humans [251]. To reach this, the postnatal phase is characterized by extensive remodeling via mitophagy, enhanced mitochondrial biogenesis, and subsequent mitochondrial maturation. Fusion and fission processes mediated by mitofusins (MFN1/2) and dynamin-related protein (DRP1), respectively, determine the mitochondrial morphology [142]. Since the transition to oxidative metabolism is a prerequisite for effective cardiomyocyte maturation, mitochondrial and cardiomyocyte maturation are intertwined.
To facilitate the metabolic transition process, fetal mitochondria are removed by mitophagy, which has been shown to be supported by the mitofusin MFN2 and the ubiquitin protein ligase Parkin [95]. A cardiomyocyte-specific ablation of Parkin has been demonstrated to block metabolic maturation [95], implying the importance of mitophagic removal for the overall maturation process of mitochondria. During maturation, mitochondrial cristae gradually acquire a more lamellar form, increasing the inner surface of mature mitochondria and thus enhancing their function [261]. Mitofusins are found to be crucial for this process as ablation of MFN1/2 results in the loss of cristae, which hampers mitochondrial biogenesis and organization [217]. In addition to the more complex inner structure, mitochondria also establish extensive networks during maturation. In mature cardiomyocytes, mitochondria are attached to the sarcoplasmic reticulum and form functional complexes for efficient substrate flux between the ATP-generating mitochondria and ADP-producing sarcomeres [258]. Mutation of ACTN2, which codes for the structural protein α-actinin 2, a core component of sarcomeres, is reported to negatively affect the size and spatial distribution of mitochondria, thereby impairing structural maturation [105].
During maturation, sarcomere length increases, while the gradual formation of Z-disks, I-, H-, A-, and M-bands attests to their improved structural organization [26, 181]. Sarcomeres, in turn, align to increasingly more organized myofibrils that exert enhanced contractile force. In humans, contractile force raises up to two orders of magnitude from neonatal individuals [314] to adults [197]. Together with the increased formation of mitochondria, sarcomere expansion is a crucial factor of cardiomyocyte maturation, not only resulting in enhanced contraction forces but also in increased cardiomyocyte sizes [106]. Interestingly, PGC1, primarily known as a regulator of energy metabolism, has also been shown to be involved in cardiomyocyte hypertrophy [199], stressing the close interconnection of metabolic and structural maturation. This maturational hypertrophy eventually yields cardiomyocytes that reach a length of around 150 µm and a volume of 40,000 μm3, more than five times larger than fetal cardiomyocytes or cultured hPSC-CMs [85, 144].
In hPSC-CMs, essential protein components of sarcomeres, such as titin, α-actinin 2, cardiac myosin-binding protein C, and myomesin, become markedly enriched upon prolonged cell culture [36]. Furthermore, sarcomere maturation is associated with a switch of respective proteins to their adult isoforms by transcriptional and posttranscriptional regulation. For example, titin switches from titin N2BA to the less-compliant N2B isoform, thereby providing increased passive cellular stiffness [167]. In mature cardiomyocytes, passive forces increase with higher beating frequency, referred to as a positive force–frequency relationship [314]. An isoform switch can also be observed for myomesin [2] and troponin (TnI), switching from the slow skeletal isoform (ssTnI) to the fast cardiac TnI isoform (cTnI) [253]. Moreover, isoforms of myosin light (MYL) and heavy (MHC) chains switch during maturation. While a switch of MYL7 to MYL2 is generally associated with cardiomyocyte maturation [65, 158], the ratio of α-MHC and β-MHC in adult hearts highly depends on the species. Smaller mammals tend to have increasing amounts of the α-isoform and larger mammals, including humans, tend to switch to the β-isoform [72, 108, 304]. Collectively, the mature isoforms provide increased contractility compared to fetal cardiomyocytes or in vitro-generated hPSC-CMs [296].
A further structural change affecting contractile function is the formation of T-tubules that occurs late in fetal development in humans and postnatally in a number of other mammals [151]. T-tubules are invaginations of the sarcolemmal membrane that facilitate signal propagation into the cell interior, thereby mediating efficient excitation–contraction coupling. Although a number of proteins required for cardiac T-tubule formation have been identified, it is still not fully understood how exactly T-tubule maturation is initiated and regulated [107]. However, it is known that their density increases severely during cellular maturation [259], which fosters the electrophysiological maturation of cardiomyocytes, as specified below.
Electrophysiological maturation
During excitation, extracellular calcium passes the cell membrane via L-type calcium channels (Cav1.1-Cav1.4), which in turn triggers the release of intracellular calcium from the sarcoplasmic reticulum via the ryanodine receptor 2 (Ryr2) [21]. Increased calcium levels then activate myofibril contraction. Subsequently, cytoplasmic calcium is cleared by the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) and the Na+–Ca2+ exchanger (NCX) [21]. The presence of T-tubules enables closer proximity of these compartments, thereby rendering calcium handling and excitation–contraction coupling more rapid in mature cardiomyocytes [259, 327]. Conversely, an absence of T-tubules in stem cell-derived cardiomyocytes results in less synchronized Ca2+ transients [171]. A positive correlation between a rise in the upstroke velocity and an increase in contraction force underpins the interplay of structural cues and electrophysiology [239].
Moreover, expression levels of the channels mentioned above and other proteins regulating calcium handling, such as calsequestrin and phospholamban (Plb), increase with maturation [176, 279]. In contrast, another type of calcium channel, called the T-type calcium channel, is typical for fetal cardiomyocytes and occurs in hPSC-CMs but is restricted to the conduction system in the adult human heart [208]. Thus, the absence of this channel is an indicator of cardiomyocyte maturity.
Another hallmark of mature cardiomyocytes is their negative resting membrane potential. The resting membrane potential is mainly established by inwardly rectifying potassium channels (Kir1–Kir7), which stabilize values around – 85 mV in adult ventricular cardiomyocytes [175, 183], while hPSC-CMs only reach values around – 65 mV [182, 323]. Notably, cardiomyocyte maturation is accompanied by downregulation of low conductance Kir2.3 and upregulation of high conductance Kir2.1/Kir2.2, which presumably contributes to the more negative resting membrane potential seen in adult cardiomyocytes [175]. In fact, overexpression of Kir2.1 renders the electrophysiological phenotype of hPSC-CM less proarrhythmic and more comparable to adult cardiomyocytes [170]. Notably, hyperpolarized membrane potentials are reported to inhibit cell cycle progression, which might consolidate the post-mitotic phenotype of mature cardiomyocytes [1].
A lower membrane potential reduces the fraction of activatable voltage-gated sodium channels (NaV1–NaV9) [101], which, together with a reduced expression of such channels [279], might cause the slower upstroke of the action potential found in hPSC-CM compared to mature cardiomyocytes [227]. Indeed, Nav1.1–Nav1.4 expression gradually raises up to fourfold in postnatal murine hearts, and electrophysiological analyses suggest them as the main drivers of the action potential in adult cardiomyocytes [115].
Moreover, hPSC-CM and immature cardiomyocytes demonstrate shorter plateau phases than mature cardiomyocytes, which is partially based on differences in the transient outward current carrying the repolarization process [52, 202, 309]. Those differences can be attributed to the composition of voltage-gated potassium channels (Kv1–Kv4), which possess distinct inactivation and recovery kinetics and thus mediate either transient outward K+ currents or delayed, outwardly rectifying K+ currents [202, 222]. In neonatal cardiomyocytes, the transient outward current is faster inactivated and recovers more slowly from this inactivation compared to adult human cardiomyocytes, which is in line with a higher level of Kv4.3 (mediating fast transient outward K+ currents) and a lower level of the regulatory subunit Kv-channel interacting protein 2 (accelerating Kv4.3 recovery and deactivation) [222, 309]. Primarily, the plateau phase is dependent on the kinetics of the L-type calcium channels, contributing to the inward current during this phase. Accordingly, altered expression of Cav1.2 components might add to the elongated plateau phase observed in mature cardiomyocytes [174, 233].
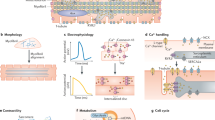
Finally, conduction velocity is higher in mature cardiomyocytes, which is thought to be mainly attributable to the improved organization of gap junctions via their accumulation at the intercalated disks in adult cardiomyocytes [102, 145]. However, besides the remodeling of gap junctions, the increased cell size of mature cardiomyocytes might substantially contribute to this effect as experimental data and modeling approaches suggest higher conduction velocities in larger cells [268, 269]. These observations further stress how electrophysiological, structural, and metabolic maturation aspects in cardiomyocytes are highly interconnected. Figure 1 provides a summary of features characterizing mature cardiomyocytes.

Hallmarks of immature and mature cardiomyocytes spanning metabolic, structural, and electrophysiological aspects
Different hormones impact cardiomyocytes via shared signaling pathways
Cardiomyocytes are exposed to a variety of hormonal cues that regulate their development and function. Together with the nervous system, the endocrine system mediates signals to induce adaptation processes to meet changing requirements depending on the developmental stage and (patho-)physiological conditions. Here, we focus on hormones impacting cardiac maturation and outline the signaling pathways involved. In fact, some pathways are shared by multiple hormones, as can be explored in Fig. 2. Moreover, cardiomyocytes produce hormones that exert paracrine and systemic effects, which will also be briefly addressed in this section.

Secretion and downstream signal transduction of hormones impacting cardiomyocyte maturation
Thyroid hormones
Thyroid hormone (TH) production is governed by the hypothalamus–pituitary–thyroid gland axis. Based on environmental and endogenous cues, the hypothalamus secretes the thyrotropin-releasing hormone, which accordingly activates the pituitary gland to release the thyroid-stimulating hormone into circulation. This hormone, in turn, stimulates follicular cells of the thyroid gland to synthesize the prohormone thyroxine (T4) and triiodothyronine (T3), which is considered the active form [24]. Plasma membrane transporters such as the monocarboxylate transporter family, the organic anion transporter family, or the L-type amino acid transporter family support the cellular uptake of thyroid hormones [24].
Intracellularly, THs can bind to two thyroid receptors (TRα and TRβ) that mediate TH signaling predominantly as ligand-dependent transcription factors [62]. For most species, TRα is the dominant form in the heart [27, 40, 89, 312]. Upon binding of T3, nuclear thyroid receptors form heterodimers with the retinoic acid receptor and interact with the so-called T3 response elements (TRE) to activate gene expression, a process referred to as the genomic pathway [62]. Some TRα isoforms are not located in the nucleus but are found in the cytoplasm, at the plasma membrane, or in mitochondria, a finding that has sparked interest in the cytoplasmic functions of this receptor [10].
Moreover, THs can bind cell surface receptors, for example, integrin αvβ3, and mediate non-genomic pathways [62]. Main signaling pathways involve, on the one hand, phosphatidylinositol 3-kinase (PI3K), protein kinase B (AKT), and mTOR [146] and, on the other hand, mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) [283]. Both signaling pathways are involved in controlling cell cycle progression and proliferation and hence regulate cardiac hypertrophy. Interestingly, thyroid hormones share these non-genomic pathways with steroids, although these initially act via distinct receptors [62]. The specific impact of thyroid and steroid signaling on cardiomyocyte maturation will be covered later in this review.
In general, the biological activity of THs is primarily modified by conversion either into the active form T3 or the more inactive forms T4, 3,3′,5′-L-triiodothyronine (reverse T3, rT3) or 3,3' diiodo L thyronine (T2), catalyzed by deiodinases. Type I and II deiodinases (DIO1, DIO2) convert T4 to T3, thereby enhancing TH signaling, while type III deiodinase (DIO3) mitigates signaling by inactivating T4 and T3 [24]. Differential expression of the involved transporters, TRs, and deiodinases allows the modulation of TH signaling on the individual cellular level.
Corticosteroid hormones
Corticosteroid hormones (CH) encompass mineralocorticoids, glucocorticoids, and sex hormones (SH), such as androgens and estrogens, which are addressed below in a separate section. All of these hormones are produced in the cortex of the adrenal gland, although the latter are primarily secreted by the testis and ovary, respectively. Secretion of mineralocorticoids and glucocorticoids is triggered by the adrenocorticotropic hormone (ACTH) released from the pituitary gland as well as angiotensin II [234, 270], which can be locally produced in the anterior pituitary gland and the adrenal cortex itself but also in the ovaries, testes, kidneys, heart, blood vessel walls, and fat tissue. Moreover, angiotensin II can be converted from its circulating precursors by the angiotensin-converting enzyme [221]. Cortisol and corticosterone are the predominant glucocorticoids in humans [203, 291] and mice [35, 286], respectively. Upon stimulation by ACTH or angiotensin II, cortisol is synthesized in mitochondria via conversion of the less active cortisone by type 1 11β-hydroxysteroid dehydrogenase [39].
Corticosteroid hormones can bind to the ubiquitous glucocorticoid receptor (GR) and the closely related, more tissue-dependent mineralocorticoid receptor. While alternative translation also results in some less-abundant isoforms, in most tissues, the GR is present in two main isoforms (GRα and GRβ) [56]. Ligand binding triggers homodimerization and translocation to the nucleus, where the GRs interact with DNA response elements (GRE) to adjust gene expression via this genomic pathway [265]. In the same way, GRs can bind to the circular DNA of mitochondria, thereby regulating mitochondrial gene transcription [232]. Moreover, steroid hormones can affect gene expression via the interaction of the GR with transcription factors, such as nuclear-factor-κB (NF-κB) and activator protein-1 (AP-1). By forming complexes with these transcription factors, GR interferes with their binding activity, thereby indirectly repressing the expression of their target genes [63].
Additionally, membrane-associated GR can mediate non-genomic pathways of steroid hormone action. These involve, on the one hand, PI3K and MAPK signaling, thereby affecting cell cycle progression and proliferation through the same pathways as thyroid hormones [62], and, on the other hand, phospholipase C, Src kinase, Ca2+/calmodulin-dependent protein kinase II, and synapsin-I, thereby affecting intracellular calcium handling and mitochondrial function [12, 252, 284].
Sex hormones
Steroid sex hormones are usually classified into three types that exert distinct functions: estrogens, androgens, and progestogens. Although these hormones are primarily categorized based on their effects on processes of sexual maturation, they have also been demonstrated to affect other aspects of organismal development, such as bone growth, metabolism, and adaptations in the cardiovascular system [149, 248].
Among other hormones, the anterior pituitary gland secretes gonadotropins, which regulate the production of female and male sex hormones in the ovaries and testes, respectively [118]. Additionally, minor amounts of sex hormones are produced in the adrenal cortex upon stimulation by ACTH, which is also released by the anterior pituitary gland. However, the main function of this hormone is to trigger glucocorticoid release, and the amounts of sex hormones produced are rather insignificant [118]. In females, estrogens, including estradiol, estrone, and estriol, can also be synthesized in the corpus luteum and the placenta. In post-menopausal women and men, circulating estrogens are predominantly converted from adrenal and ovarian or testicular androgens, respectively [118]. Androgens, including testosterone, androstenediol, and androsterone, are mainly synthesized in the testes but are also produced in the ovaries and the adrenal cortex of both genders, as mentioned before.
The sex hormone-binding globulin (SHBG) regulates the levels of free sex steroids that can easily enter the cell by passive diffusion. The “free hormone hypothesis,” which claims that levels of free hormones predominantly determine the biological activity of a given hormone [187], applies only to a limited extent for sex steroids. In fact, it has been demonstrated that cellular uptake of SHBG-bound androgens and estrogens could be realized via endocytosis and that sex-steroid signaling, at least in part, depends on this route [114].
Estrogens can bind to three forms of estrogen receptors [ERα, ERβ, and G-protein-coupled estrogen receptor (GPER)]. In the nucleus, estrogen binding to ERs triggers the recruitment of coactivators that facilitate binding to DNA via estrogen response elements (ERE). Hence, the ER acts as a ligand-gated transcription factor, mediating the effects of estrogens via the genomic pathway [154]. Like GRs, ERs can also bind to mitochondrial DNA and induce transcription of mitochondrially encoded genes directly [232] or indirectly via the nuclear respiratory factor-1 that promotes transcription of the mitochondrial transcription factor Tfam, which regulates the mitochondrial-encoded gene expression [185]. Similarly to thyroid hormone signaling, ERs located in the plasma membrane or cytosol [243] can mediate more rapid effects via non-genomic pathways, such as PI3K/AKT/mTOR and MAPK/ERK, which are implicated in proliferation and hypertrophy as well as calcium cycling and eNOS synthesis [248].
In the same way as estrogens, androgens cross the cell membrane and primarily bind to nuclear androgen receptors (ARα and ARβ), thereby inducing the genomic pathway via DNA response elements in target gene promoters. Testosterone can be converted intracellularly to dihydrotestosterone, which forms much more stable complexes with ARs and thus can amplify the androgen signaling in individual cells [315]. Membrane-bound receptors mediate non-genomic effects most prominently via the activation of protein kinase A (PKA) and protein kinase C (PKC), thereby impacting calcium cycling but also via activation of MAPK/ERK, affecting proliferation and hypertrophy [76].
Progesterone binds to two isoforms of nuclear progesterone receptors (PR-A and PR-B), which initiates the genomic pathway by dissociating the receptor from a chaperone complex, thereby allowing homodimerization and interaction with progesterone response elements [248]. The binding of DNA can either decrease or increase gene transcription, depending on the recruitment of coactivators or RNA polymerase II to the initiation site. Three membrane-bound progesterone receptors (mPRα, mPRβ, and mPRγ) act as G-protein-coupled receptors and decrease 3′,5′-cyclic adenosine monophosphate (cAMP) synthesis upon ligand binding, thereby inducing non-genomic actions [248]. Generally, both female and male sex hormones can act via genomic and non-genomic mechanisms.
Igf system
The Igf system includes two forms of insulin-like growth factors (Igf1 and Igf2), their receptors, and Igf-binding proteins (Igfbps) that regulate the bioavailability of Igfs. Igf1 is primarily produced in the liver in response to growth hormone (Gh) secreted by the pituitary gland but can also be generated in other organs such as the heart [67].
Because Igfs share some structural homology with insulin, they can bind either to the Igf1 receptor (Igf1R) or the insulin receptor (IR). In contrast to the hormones presented here before, Igf1 does not directly mediate a genomic pathway. The Igf1R belongs to the receptor tyrosine kinase family and triggers phosphorylation of intracellular lipids, second messengers, and serine/threonine kinases upon ligand binding, thereby activating non-genomic pathways such as PI3K/AKT/mTOR and MAPK/ERK [288]. Besides these classical Igf1 signaling pathways, it has been shown recently that Igf1 can also activate phospholipase C (PLC) via a heterotrimeric G protein, initiating an inositol 1,4,5-trisphosphate (IP3)-dependent signaling pathway that eventually affects intracellular calcium levels [126].
Igf2 can bind Igf1R and the mannose 6-phosphate receptor with high affinity, which is, therefore, also known as the Igf2 receptor (Igf2R) [58]. In contrast to Igf1R, this receptor lacks the tyrosine kinase domain. Instead of inducing phosphorylation cascades, binding this receptor results in internalization and degradation of the ligand [206] and thus inhibits the signaling of Igf2 via the Igf1R [71]. Accordingly, the regulation of Igf signaling can be considered the predominant role of this receptor.
Cardiomyocyte-derived hormones
The heart is not only the target organ for many hormones but also a site of hormone production, as extensively reviewed by Chiba et al. [45] and shortly outlined in the following. The most prominent cardiac hormones are ANP, which is produced by atrial cardiomyocytes [28], and the structurally and functionally related BNP, which has been originally discovered in the porcine brain [273] but is predominantly secreted from the ventricle [196]. ANP and BNP are stored in secretory granules and continuously released through the classical secretory pathway. Stimulation by mechanical forces (stretch) or agonists, such as endothelin1 and phenylephrine, can transiently increase their release [32]. Endothelin1 itself is also produced in cardiomyocytes [277], although vascular endothelial cells are the main source of this hormone.
Circulating ANP and BNP binds to three natriuretic peptide receptors (NPR1-3). NPR-1 and NPR-2 contain a guanylate cyclase domain, thus increasing intracellular cyclic guanosine monophosphate (cGMP) levels upon ligand binding [47], while NPR-3 lacks the guanylate cyclase domain and serves as a clearance receptor. Increased cGMP levels stimulate the cGMP-dependent protein kinase G (PKG) that phosphorylates a wide number of downstream targets [281]. For example, PKG phosphorylates Plb, thereby activating the associated Ca2+-ATPase SERCA-2a and enforcing calcium sequestration. Additionally, PKG phosphorylates IP3 receptor-associated PKG-I substrate, which inhibits the release of Ca2+ mediated by the inositol trisphosphate receptor. On the cellular level, the cGMP–PKG signaling pathway mediates the regulation of relaxation and contraction, as well as hypertrophy and apoptosis [281]. Systemically, ANP and BNP trigger natriuresis and affect the perception of satiety [28, 98, 300].
Other hormones are not predominantly produced in the heart, yet cardiomyocytes contribute to their systemic levels. For example, cardiomyocytes might even influence bone metabolism via osteocrin [46] or parathyroid hormone-like protein [64], and affect body growth via growth differentiation factor 15 [307]. However, most cardiac hormones act in an autocrine or paracrine manner to regulate local blood pressure and angiogenesis, proliferation and hypertrophy, metabolism, as well as inflammation [45]. Such cardiomyocyte-derived hormones are, for example, vasostatin1, follistatin-like 1, fibroblast growth factor 21, and phospholipase A2. Moreover, paracrine signaling by calcitonin is shown to prevent atrial fibrosis and fibrillation [172], and cardiac-derived oxytocin [132] might convey protection from ischemia–reperfusion injury [97].
Thyroid hormones govern various aspects of cardiomyocyte maturation
Levels of circulating THs are low in postnatal mice and rats but display a dramatic increase in the first days of life [79, 201]. This increase in TH levels is reportedly associated with a preadolescent burst of cardiomyocyte proliferation [201]. However, employing different means to verify these findings, other groups have not observed such a burst [5, 267], but rather have confirmed older studies, stating that postnatal cardiomyocytes respond to administered TH by rapid growth in volume but not in number [209]. Such opposing results point to experimental limitations in tracking cell proliferation in vivo and emphasize the need for more sophisticated approaches. While their effect on postnatal proliferation is still under debate, evidence for a crucial role of TH in prenatal cardiomyocyte maturation accumulates.
Studies in thyroidectomized and T3-infused sheep suggest that increased TH levels promote cardiomyocyte maturation in terms of improved calcium handling, increased cell size, and binucleation [41]. In line with that, Van Dusen et al. have identified TRα as upregulated in mature cardiomyocytes compared to Myh7-positive immature cardiomyocytes [294]. Of note, in the same study, cardiomyocyte maturation is demonstrated to be epigenetically regulated by RNF20/40, which is reported to be a transcriptional coactivator for PPARγ [133]. Although another study in chicken embryos could demonstrate neither increased cell size nor elevated binucleation rates of cardiomyocytes treated with T3 for 24 h [278], other studies support the idea that T3 promotes cardiomyocyte maturation. For example, T3 treatment is reported to foster cardiac differentiation of murine embryonic stem cells (mESCs) [164] and drive maturation of hPSC-CM influencing basal beating rate, the upstroke of the action potential and mitochondrial metabolism [117], as well as increasing sarcomere length and force generation [319].
Although T3 is shown to repress cardiac Plb gene expression via epigenetic mechanisms [18], several studies demonstrate that T3 improves calcium handling overall by inducing increased expression of the calcium ATPase SERCA and the calcium channel Ryr2 via the classical genomic pathway [117, 129, 164, 319], thereby enhancing contractile function. Similar results are obtained when supplementing engineered cardiac tissues from neonatal rats [147] or human-induced stem cells [229] with T3, which also improved contractile properties.
Later, this approach has been refined by adding a low-frequency pacing protocol, which conveys synergistic effects [131]. Nevertheless, T3 seems to be the main driver of a shortened cardiac action potential and maturation of sarcomeres, resulting in the enhanced contractile force. Referring to the induced physiological hypertrophy as well as improved sarcomere organization and force generation, the authors claim that the structural and functional characteristics of their engineered myocardium match those of the adult ventricle [131]. However, some features still resembled immature cells, such as a random distribution of the gap junction protein Cx43 and a rather small cell size, demonstrating that fully matured cardiomyocytes are still to be achieved.
As outlined before, physiological hypertrophy of cardiomyocytes is accompanied by structural changes of sarcomeres, including isoform switching of involved components, such as alpha-myosin heavy and light chain, as well as titin and troponin. It has long been known that thyroid hormones influence the switch from β-MHC to α-MHC [48, 110], which later has been demonstrated to be epigenetically regulated [111]. A lack of β-MHC to α-MHC conversion is observed in the offspring of mice with ablated thyroid glands [290], stressing the importance of prenatal exposure to thyroid hormone. For titin, T3 has been shown to trigger the isoform switch from N2BA to N2B in cell culture [157], while reduced T3 levels increase the ratio of N2BA to N2B, resulting in reduced forces in cardiomyocytes of hypothyroid rats [316]. Similar results are obtained for the troponin isoform switch from ssTnI to cTnI, demonstrating an accelerated transition to cTnI in rodent cardiomyocytes treated with T3 [16].
On the other hand, when hPSC-CMs are exposed to T3, no such effect on troponin isoform switch could be observed [16]. These results should serve as a reminder of the potentially very limited transferability of results from rodent models to human patients. Also, another sarcomere protein, the so-called M protein myomesin 2, is demonstrated to be downregulated in rat ventricles after daily injections of T3 [244]. This observation implies that the effects of T3 on different sarcomere components are not necessarily consistent.
Physiological hypertrophy of cardiomyocytes is not only due to sarcomere elongation but also caused by increased mitochondrial biogenesis. T3 [282] as well as T4 [93] are shown to induce the expression of genes that promote mitochondrial biogenesis via the non-genomic pathway. In particular, elevated PPARα, PGC1α, and Tfam levels are identified as underlying regulators of the increased mitochondrial content and function observed in thyroxine-stimulated cardiomyocytes [93, 209, 229, 317]. In line with that, the expression levels of many mitochondrial genes are reduced in mice lacking cardiac TRα [119]. Accordingly, mutant hearts demonstrate impaired mitochondrial biogenesis, resulting in cardiomyocytes with a 47% reduction in size, but, on the other hand, an increased number of proliferative diploid cardiomyocytes. This observation puts thyroid hormones at the crossroad of proliferation and maturation.
As outlined before, the transition from proliferation to maturation is also marked by changes in the electrophysiological profile of cardiomyocytes. Early studies in knock-out mice demonstrate that thyroid signaling affects intrinsic heart rate regulation [135], suggesting a link between thyroid signaling and electrophysiological maturation of cardiomyocytes. T3 treatment is shown to alter the expression levels of cyclic nucleotide-gated channels HCN2 and HCN4 [83, 89, 129] and to enhance the respective I(f) current in pacemaker cells [83, 275], while this current is decreased in T3 + dexamethasone-treated hPSC-CMs [305].
In hPSC-CMs, T3 + dexamethasone treatment results in a more hyperpolarized resting membrane potential, faster maximum upstroke velocity, and higher conduction velocity but also shorter action potential duration [305]. Shortened action potential durations in T3-treated cells are attributed to an increased Na + –Ca2 + exchanger activity [275] and an enhanced inwardly rectifying potassium current [245, 305]. In line with that, further knock-out studies in mice reveal the voltage-gated potassium channel Kv 4.2 as regulated by thyroid receptors via the genomic pathway [89]. On the other hand, acute T3 treatment is demonstrated to prolong the action potential duration by slowing down the inactivation of sodium channels [54], while another study reports that T3 could induce bursting of cardiac sodium channels by directly acting extracellularly at the channel and independently of nuclear transduction [70]. These results point to the complexity of thyroid signaling via genomic and non-genomic pathways and illustrate how direct and indirect actions might mediate distinct effects.
Similarly inconsistent are findings on TH effects on conduction velocity. Mice carrying a mutant thyroid hormone receptor demonstrate a decreased atrial conduction velocity [7], thus suggesting a link between thyroid signaling and the organization of gap junction proteins, such as connexins Cx40 and Cx43. Indeed, thyroid hormone levels are reported to be positively correlated with Cx40 mRNA abundance [7] and negatively correlated with Cx43 mRNA abundance [13]. However, although there are thyroid hormone response elements in the Cx43 promoter, earlier studies using the same rat model did not find a significant effect of thyroid stimulation on Cx43 gene expression [272]. Interestingly, in the study reporting decreased Cx43 mRNA levels upon T3 treatment, these are shown to be accompanied by a decreased conduction velocity in diabetic rats and increased conduction velocity in non-diabetic rats [13]. This reinforces the notion of a multifactorial network that seems to underlie and modulate thyroid signaling effects.
Corticosteroids function synergistically to support the maturation of cardiomyocytes
As described above, thyroid and steroid hormones share non-genomic pathways and thus have overlapping effects on cardiomyocyte maturation. However, these hormones are not necessarily redundant but can mediate synergistic effects. For example, only the combined administration of T3 and the glucocorticoid dexamethasone induced T-tubule formation in hPSC-CM [220]. In fact, glucocorticoids, such as cortisol, can alter the thyroid hormone metabolism via DIO2/3 and are demonstrated to be relevant for the increase of fetal plasma T3 levels near birth [257]. Interestingly, a study in chicken demonstrates that effects of glucocorticoids on T3 and T4 levels are dependent on the developmental stage [59], hinting at further layers of regulation.
Similarly to thyroid hormones, levels of circulating glucocorticoids dramatically increase toward term [78, 263]. This increase is primarily established by an enhanced glucocorticoid generation in the fetal adrenal gland but can be fostered further by downregulation of circulating binding globulins [78, 263]. Glucocorticoids are crucial for the maturation process of fetal organs and support postnatal survival, which has led to their clinical application in humans [191]. In the heart, glucocorticoid signaling supports the structural and functional maturation of cardiomyocytes, as outlined below.
For example, maternal glucocorticoid treatment, as clinically applied in humans, promotes the binucleation of cardiomyocytes and improves cardiac function in preterm piglets [152]. Although the authors could not detect an increase in myocyte volume or sarcomere length 48 h after glucocorticoid exposure, proliferation and apoptosis are more similar to term hearts, suggesting ongoing remodeling processes eventually resulting in structural maturation. Indeed, several studies in rats demonstrate increased cardiomyocyte length and volume, as well as actin and myosin heavy-chain content, when data have been collected 1–50 weeks after neonatal glucocorticoid exposure [15, 161, 301]. Interestingly, another study in fetal sheep has been able to demonstrate an effect of glucocorticoid exposure on cardiomyocyte volume even within 56–72 h. Nevertheless, no effect on binucleation was observed [180], which opposes the results in piglets. This emphasizes the inter-species differences that restrict the transferability of particular results. However, an impact of glucocorticoids on the morphological features of cardiomyocytes has been noted in a variety of model organisms, and the concordance of in vitro and in vivo studies contributes to the validity of a link between glucocorticoids and structural maturation of cardiomyocytes. As such, glucocorticoid treatment is shown to enhance the assembly of sarcomere Z-disks and myofibrils in vitro, which results in improved contractility in murine fetal cardiomyocytes [240] and ESC-derived cardiomyocytes [326]. Conversely, GR knock-out mice demonstrate decreased contractile function due to the formation of short, disorganized myofibrils and impaired calcium handling [241]. Moreover, glucocorticoid treatment induces the generation of respective proteins relevant for calcium handling, such as Cav1.2, Ryr2, SERCA, and NCX [240], while GR knock-out mice demonstrate decreased expression levels of these genes [241]. Additionally, studies in sheep demonstrate that glucocorticoids support electrophysiological maturation by enhancing the gene expression of cardiac sodium channels [74].
Finally, metabolic maturation is improved by corticosteroid hormones. Although the mitochondrial morphology of cardiomyocytes seems not to be affected by steroids like dexamethasone, it does promote Parkin-mediated mitophagy in ESC-derived cardiomyocytes, thereby supporting the perinatal switch to fatty acid oxidation [326]. Glucocorticoid effects on metabolic maturation are mediated by two main factors, namely PPARγ and PGC1α. Antenatal dexamethasone administration has been shown to increase the expression of PPARγ and creatine kinase, which serve the rapid regeneration of ATP and intracellular energy transport [193]. At the same time, PGC1α is essential for the induction of mitochondrial gene expression and the increase of cellular respiration observed in glucocorticoid-treated cardiomyocytes [240, 326].
However, a recent study reports that antenatal glucocorticoid treatment decreased PGC1α and GR expression in a sheep model of preterm birth [130], suggesting that exogenous glucocorticoids might potentially also interfere with cardiomyocyte maturation by downregulation of GR. Additionally, the study of Kim et al. on the effect of maternal glucocorticoid treatment on cardiac maturation in preterm piglets demonstrates clear sex differences [152]. Interactions of the different corticosteroid hormones, including sex hormones, should be taken into account when clinically applying antenatal glucocorticoids in humans. Accordingly, timing and dosage should be considered precisely on the individual level.
Sex hormones affect cardiac metabolism and contractile function
As mentioned earlier, there are significant sex differences in cardiovascular health and function, which are largely attributed to the action of sex hormones. Besides well-known cardioprotective effects of estrogens that will be covered later, some studies also hint at the impact of sex hormones on the electrophysiological maturation of cardiomyocytes. For example, the calcium-handling proteins SERCA-2a, NCX1, and Cav1.2α are more abundant in females [51, 218], and studies in ovariectomized rats underpin the association of their abundance and estrogen levels [50]. While SERCA, as well as Plb and Ryr, seem to be less affected by estrogen levels, the abundance of NCX1 and Cav1.2α is strongly regulated by estrogen [50], as also demonstrated in female hPSC-CM [218]. Intriguingly, in male hPSC-CM, estrogen fails to induce these effects [218]. On the other hand, testosterone induces a similar effect on these calcium-regulating proteins in isolated ventricular rat myocytes [91], which also show enhanced contractility upon testosterone treatment [92].
In line with the aforementioned increased abundance of Cav1.2α in female hearts, the L-type calcium current is accordingly larger than in males [298]. Transient outward K+ currents, instead, tend to be smaller in females compared to males [298]. Utilizing computer simulations, Verkerk et al. explored the potential functional effects of these gender-dependent differences. They report that the slightly larger depolarizing calcium current, together with slightly smaller repolarizing potassium currents, results in significantly longer action potentials and greater susceptibility to abnormal depolarizations in female cardiomyocytes [298]. Although other parameters, such as resting membrane potential and upstroke velocity, show no gender disparities [298], the prolonged action potential in female cardiomyocytes suggests an electrophysiologically slightly more mature phenotype, which is in line with the beneficial effect of female sex hormones on calcium handling.
However, data on the impact of estrogens on contractile function are conflicting. Ovariectomy has been reported to exert negative [211, 250, 289], positive [57, 139, 156, 224], or no effects [50, 310] on contractile function, rendering evaluation of the underlying mechanisms complicated. A reason for these conflicting data, as well as the respective disparities concerning the effect of estrogen replacement, might reside in the experimental design with respect to the time point of functional measurements. For example, Paigel et al. demonstrate that results differ when myocardial contractility is assessed 7 or 60 days after ovariectomy [210]. Although experimental outcomes differ across studies, the data suggest a contribution of AMPK [289] and PKA [156] in mediating the estrogen effects on the L-type calcium current and contractility. Further comprehensive studies thoroughly considering actual hormone levels and the time course of events are needed to elucidate the effects of sex hormones on contractile properties of cardiomyocytes and the mechanisms underlying these effects.
ERRs that share sequence homology with ERs [88] are not only involved in energy homeostasis [87] but also modulate various aspects of metabolic, structural, and electrophysiological maturation of cardiomyocytes. In particular, ERR knockdown in hPSC-CMs results in smaller mitochondria with less and disorganized cristae, a hampered isoform switch of troponin and myosin regulatory light chain, as well as reduced expression of ion transporters and their subunits, such as Plb, Ryr2, Atp2a2, Atp1a1, Atp1a2, Cacna2d3, and Kcnq1 [247]. Conversely, a recent study utilizing an ERRγ agonist demonstrates an increased oxygen consumption rate and ATP production, larger cell size, and longer sarcomere length in the presence of T-tubules, as well as increased action potential amplitudes, upstroke velocity, and maximum conduction velocities in treated hPSC-CMs [189]. Although this orphan receptor does not bind estrogens and natural ligands have yet to be discovered, ERRs can influence estrogenic signaling via crosstalk [86, 87], which is why they are mentioned here.
While the major hallmarks of cardiomyocyte maturation apply to both sexes, mitochondria demonstrate a clear sexual dimorphism, which is thought to originate from the exclusive maternal inheritance of their genome, resulting in an optimized function in female compared to male individuals [287]. This contributes to gender differences in cardiac function, as well summarized by Ventura-Clapier et al. [297]. Female cardiomyocytes seem to have fewer but more efficient mitochondria regarding fatty acid utilization, oxidative capacity, and ATP production [297, 299]. As mentioned before, estrogens can induce the transcription of mitochondria-encoded genes relevant for oxidative phosphorylation via the genomic pathway downstream of ERs [185, 232]. In this way, estrogens can enforce the metabolic switch during cardiomyocyte maturation. Additionally, studies in ovariectomized rats reveal that estradiol could also increase oxidative capacity and decrease oxidative stress by binding the membrane-bound GPER that subsequently activates MAPK signaling [249].
Interestingly, sex differences in the gene expression profiles of cardiac cell populations are indeed most pronounced in cardiomyocytes [264]. While these differences can be observed across all stages of development, most of them become more pronounced in adult individuals. The insightful transcriptome and chromatin accessibility study of Sim et al. reveals that cardiomyocyte maturation is associated with increased accessibility at sites of the glucocorticoid response element (GRE), the androgen response element, and the progesterone receptor. Additionally, open chromatin regions in female cardiomyocytes are highly enriched for AP-1-JUN motifs. Applying extracellular field potential and impedance measurements, Sim et al. further demonstrate a positive inotropic effect of progesterone, which increases contractile force, upstroke velocity, and relaxation velocity in human embryonic stem cell-derived cardiomyocytes. Interestingly, there are no sex differences in gene expression levels of the PR and AR, which are both upregulated during cardiomyocyte maturation [264].
However, in contrast to the various studies on estrogen effects, the number of studies on androgens and the AR during cardiomyocyte maturation is by far less. The AR is expressed in neonatal and adult cardiac tissue in rodents as well as in humans of both genders [184, 226], where it mediates a hypertrophic response to testosterone [184] through ERK and mTOR signaling [8]. Notably, consequences of this hypertrophic response are reported to depend on the exposure duration. While short-term exposure to testosterone increased myocyte contractility and ejection fraction, long-term exposure for 12 weeks affected these parameters negatively, thereby implying pathological hypertrophy [303]. This underlines the necessity of tight regulation of hormone actions in time and dosage.
Goldman-Johnson et al. report an impact of testosterone on cardiomyocyte differentiation. Particularly, they observe an increased formation of α-actinin and tropomyosin-positive cardiomyocytes from murine embryonic stem cell lines upon testosterone exposure [94]. Later, this finding has been substantiated by the finding that testosterone stimulates transcription factors, such as GATA4, MEF2C, and Nkx2.5, via the genomic pathway [3]. However, studies in ovariectomized rats have not identified any acute or chronic effects of testosterone on hallmarks of structural or functional maturation with respect to contraction dynamics, calcium cycling, or MHC isoform switch in adult individuals [17]. In line with that, another study in sheep demonstrates a restriction of a positive effect of testosterone on cardiomyocyte proliferation and maturation regarding binucleation to a specific period early in pregnancy [137]. Interestingly, insulin-like growth factor 1 signaling seems at least partly to mediate the observed effects [137].
IGF triggers cardiac hypertrophy and affects oxidative metabolism
Just like for thyroid and corticosteroid hormones, early studies demonstrate a quick rise in systemic Igf1 concentrations after birth [60, 90]. Clinical studies suggest that thyroid hormones directly modulate Igf1 levels [127], and more recently, in vivo and in vitro studies confirm that T3 increases the expression of Igf1 as well as Igf1R in cardiomyocytes, eventually resulting in enhanced phosphorylation of PI3K and AKT protein [322]. Both Igf1 and T3 stimulate the phosphorylation of AKT and ERK in cardiomyocytes. However, intriguingly, a combination of both hormones demonstrates distinct effects depending on the maturational state of the cells, reducing phosphorylation in early phases and increasing phosphorylation in later phases of fetal cardiac development [42]. Early in vitro studies report that Igf1 signaling promotes cardiomyocyte proliferation but not cellular hypertrophy [138], which later has been confirmed in fetal sheep cardiomyocytes [276]. However, other in vivo studies utilizing cardiomyocyte-specific Igf1R overexpression suggest that Igf1 indeed triggers physiological hypertrophy [186], thus implying a role of Igf1 in structural maturation.
In rats treated with Gh, a steady increase in serum Igf1 levels is accompanied by an increase in myofiber length, total cardiomyocyte volume, and the total number of cardiomyocyte nuclei in the left ventricle, although it was not assessed whether the latter was due to an increase in total cell number or binucleation rates [31]. Moreover, treatment with Igf1 is not only shown to increase the cross-sectional area of cardiomyocytes but also to enhance the expression of MYL2 and the troponin isoform switch from ssTnI to cTnI [61, 128], thereby promoting the assembly of myofibers [31, 69]. Yet, a positive effect of Igf1 on structural maturation is at least questionable as another study by Laustsen et al. reports an upregulation of contractile proteins, especially of the Z disk, as well as β-MHC in insulin receptor and Igf1R knock-out mice [162]. Notably, the altered gene expression of sarcomere components interferes with normal cardiac function in these mice.
Again, an explanation for the contradicting findings may lie in the time point of the readout. In fact, an early study in mice shows that local overexpression of Igf1 does result in physiological cardiac hypertrophy in early life; however, this progresses to pathological hypertrophy accompanied by decreased contractile function at later time points [66]. Myofibers that are first only hypertrophic become increasingly disorganized upon persistent Igf1 exposition, which aligns with the later findings of Laustsen et al. [162]. Thus, a precisely timed exposure of cardiomyocytes to Igf1 seems to be critical for the maturation process. On the other hand, the contractile function is also hampered in Igf1-deficient dwarf mice, which demonstrate some dysfunctions in excitation–contraction coupling [237]. Interestingly, a study in aging mice demonstrates that chronic cardiac-specific Igf1 overexpression could attenuate aging-associated contractile dysfunction by improving calcium handling [169].
Interestingly, the study of Laustsen et al. does imply a relevance of Igf1 for metabolic maturation, as Igf1 knock-out mice demonstrate downregulation of genes from the electron transport chain and mitochondrial oxidative phosphorylation pathways and, thus, a more immature metabolic profile [162]. The energy metabolism of cardiomyocytes is also shown to be obstructed in chicken models of Igf1 knockdown [96]. Conversely, Igf1 promotes the expression of the medium-chain acyl-CoA dehydrogenase and the muscle-type carnitine palmitoyltransferase I downstream of PPARα, thus fostering a more mature metabolic profile [194]. In this study, the oxidation of fatty acids was not enhanced by Igf1 administration. However, the cells were exposed to Igf1 only for 48 h. Therefore, long-term effects on oxidative phosphorylation and metabolic maturation cannot be ruled out.
When applying a cocktail of Igf1, TH, and dexamethasone for seven days in hPSC-CMs, either cultured as monolayers or in 3D cardiac microtissues, various aspects of metabolic, structural, and electrophysiological maturation are enhanced [124]. In particular, treatment with this hormone cocktail increases expression levels of PPARα and PPARγ as well as the phosphorylation of AKT and mTOR, thus supporting the transition of the metabolic phenotype toward mitochondrial oxidative metabolism. In line with that, treated cells show a higher density of elongated mitochondria attached to the sarcomeres. Structural maturation is demonstrated by a larger cell area, increased expression of sarcomeric genes such as MYH6 and ACTN2, as well as the presence of longer sarcomeres with improved alignment and T-tubules. Decreased MYL7 expression and increased MYL2 expression, together with decreased ssTnI expression, implicate an accelerated isoform switch in treated cardiomyocytes compared to the untreated control. Higher mRNA and protein levels of calcium transporters, such as Ryr2, Atp2a2, and Slc8a1, imply improved calcium handling, manifesting in faster Ca2+-transient kinetics in treated cardiomyocytes. Further indicators of electrophysiological maturation are the upregulation of high conductance Kir2.1, increased expression of Cx43, as well as a polarized gap junction distribution, resulting in a higher conduction velocity in treated cells. Taken together, the combined administration of Igf1, TH, and a synthetic glucocorticoid severely accelerates the overall maturation process in hPSC-CMs.
Another group combined a similar cocktail of Igf1, TH, and dexamethasone with a HIF-1α inhibitor and an agonist of PPARα to foster hPSC-CMs’ maturation [84]. The study demonstrates enhanced expression of genes involved in fatty acid oxidation, increased mitochondrial content and maturation as well as faster calcium transient kinetics, and higher contractility. Altogether, these studies underscore not only the interlinkage of metabolism and cardiomyocyte maturation but also that only the holistic interplay of diverse signals may be sufficient to achieve the ultimate goal of fully mature cardiomyocytes. Table 1 provides an overview of studies in the most common model organisms that involve a hormone treatment and their effects on cardiomyocyte maturation. Although some studies fail to demonstrate a beneficial effect of hormone treatment on the maturation process, most studies suggest that mimicking endocrine cues can indeed foster the maturation of cardiomyocytes and thus should be implemented in protocols to generate cardiac cells for drug testing and disease modeling.
Hormone-based strategies for cardiac regeneration
For cellular disease model systems, preclinical in vitro drug testing systems, as well as for basic research, adult-like cardiomyocytes are of utmost importance to improve the transferability of study results to clinical settings. However, while the promotion of cardiomyocyte maturation is a major goal in tissue engineering approaches in vitro, terminal mammalian cardiomyocyte maturation in vivo has been shown to pose a barrier to regeneration [230], restricting regenerative capacity to a small time window in postnatal hearts. In line with that, early inactivation of TH signaling, which usually promotes cardiomyocyte maturation, is found to prolong this regenerative window in mice [119]. During this period, Igf2 acts as a paracrine mitogen [260]. Igf2-deficient neonatal mice demonstrate impaired regenerative capacity, which can be restored by various means to increase the proportion of mononuclear diploid cardiomyocytes [260] that are drivers of heart regeneration, as mentioned before [225].
Interestingly, while being barely detectable in adult healthy mouse heart tissue, Igf2 is reexpressed upon conditions of ischemia and reperfusion, e.g., during myocardial infarction (MI) [260], which is in line with findings in zebrafish, where the ortholog of Igf2 (igf2b) is found to be upregulated during regeneration [125]. However, although the reliance on Igf signaling appears to be conserved, the involved receptors and pathways seem to differ across species. While inhibition of Igf1R signaling impairs myocardial regeneration in zebrafish [125], postnatal regeneration in mice is shown to be exclusively dependent on the insulin receptor pathway [260]. In humans, data on postnatal functions of Igf2 are scarce; however, a recent case–control study reports an association of high serum Igf2 levels with lower mortality from heart failure, implying a role of Igf2 in cardioprotection [73]. Conversely, levels of Igf2R, which acts as an inhibitor of Igf signaling, are found to be higher in heart failure patients compared to individuals without heart disease [311].
In fact, Igf1 is known to convey cardioprotective actions [274] and is even demonstrated to allow for a prognostic assessment of heart failure risk. For example, healthy participants of the Framingham Heart Study with serum Igf1 levels below the median value have had a risk of developing heart failure twice as high as individuals with Igf1 levels above the median value [295]. Also, after the onset of heart failure, Igf1 has been shown to mediate beneficial effects. High total Igf1 levels immediately after MI are associated with better clinical outcomes concerning myocardial remodeling processes and ventricular function [163].
Regarding approaches to foster heart regeneration, local myocardial Igf1 delivery with biotinylated peptide nanofibers is shown to improve cell therapy for MI in rats [61]. Also, in adult mice, short-term Igf1 treatment after MI conveys positive effects on cardiac function, scar size, and capillary density by modulating the immune cell response in the acute inflammatory phase [116]. In line with the findings on Igf2 signaling during postnatal heart regeneration in neonatal mice, the inactivation of Igf1R in cardiomyocytes does not abrogate the protective effect of Igf1 [116]. However, when Igf1R is knocked out in myeloid cells, the positive effects of Igf1 treatment on cardiac regeneration and function are depleted. Yet, Igf1 effects on cardiac regeneration are not limited to immune modulation, since it is known that Igf1 reduces apoptosis [274] and decreases reactive oxygen species (ROS) generation [9], which is another main factor underlying ischemia–reperfusion injury.
The large number of promising preclinical studies has led to the first clinical trial on the safety and efficacy of intracoronary Igf1 infusion in MI patients, the “RESUS-AMI” study [38]. This study has failed to demonstrate a significant efficacy on the level of the chosen primary readout (left-ventricular ejection fraction). However, the results point to a beneficial effect on remodeling and later preclinical studies, administering much higher concentrations of Igf1 in a porcine MI model, have demonstrated not only decreased myocardial fibrosis but also increased left-ventricular ejection fraction [14]. Upstream of Igf1 and Igf2, Gh is also shown to mediate positive effects in heart regeneration. Early studies in rats show that Gh impacts the remodeling process after MI by enhancing physiological cardiomyocyte hypertrophy and reducing adaptive fibrosis, which results in improved cardiac function [103]. Moreover, other studies demonstrate that even four weeks after MI, intracellular calcium transients and contractile function could be improved via long-term treatment with Gh, which might at least partially be attributed to the increased protein levels of SERCA-2 upon treatment observed in this study [280]. An upregulation of Gh and Igf1 and subsequently increased phosphorylated AKT levels are also suggested to underlie the cardioprotective effect of ghrelin treatment [109] in cardiomyocytes undergoing hypoxia and reoxygenation [178]. Surprisingly, the growth hormone-releasing hormone (GHRH) agonist JI-38 is reported to promote cardiac repair after MI, independently of Gh or Igf1 [141], suggesting a direct signaling pathway of GHRH.
As mentioned before, the IGF1/PI3K/AKT signaling pathway in cardiomyocytes is also activated by T3 [322], and a recent study in mice reports that a protective effect of T3 against post-MI dysfunction is based on the activation of this axis. In particular, T3 administration results in a smaller infarct area as well as reduced apoptosis and fibrosis, overall improving the cardiac function after MI, while additional injection of an Igf1R inhibitor averted these positive effects [321]. Before, a number of studies in rats has demonstrated similar beneficial effects of T3 treatment under conditions of ischemia and reperfusion. These effects are predominantly attributed to non-genomic signaling of the TRα1 receptor [214], potentially inducing signaling via AKT [44], p38 MAPK [215], JNK [213], heat stress proteins [212, 216], or involving mitochondrial biogenesis [77] or mitophagy [23].
In humans, abnormal thyroid function is associated with increased mortality in heart failure patients [192], and clinical trials analyzing the safety and efficacy of T3 treatment in such patients were already performed at the end of the last century [113]. Moreover, positive effects on hemodynamic performance are reported for intravenous administration of T3 during coronary artery surgery [236], and some clinical advantage of perioperative T3 supplementation is observed in infants undergoing cardiopulmonary bypass [231]. Another clinical trial on the effects of T3 in ischemic heart failure started in December 2022 (NCT05384847), demonstrating the continuing confidence in the effectiveness of such treatment.
These positive effects of T3 administration on heart regeneration somewhat oppose the finding of Hirose et al. that the inactivation of TH signaling improves heart regeneration in adult mice [119]. Generally, it is assumed that the presence of immature cardiomyocytes confers regenerative capacity. However, it has been shown that mature cardiomyocytes can reenter the cell cycle after dedifferentiation, which contributes to newly formed cardiomyocytes usually found in the border zone of infarcted hearts [308]. In fact, a recent study reports that experimentally induced dedifferentiation by overexpression of Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc) could mitigate cardiac damage and improve heart function after MI [43]. Potentially, thyroid hormone signaling might be crucial to facilitate the redifferentiation of newly formed cardiomyocytes to restore cardiac function and thus improve heart regeneration.
As mentioned before, clinical and epidemiological studies reveal significant sex differences in cardiac health and the outcome of cardiovascular diseases (CVDs). Data from the world health organization mortality database [29] and extensive cohort studies, such as the Framingham study that comprises data on the incidence and prognosis of CVDs over 50 years [143], clearly show that the risk of developing CVDs is several times lower in women before menopause than in men but evens out after menopause. After menopause, higher estradiol levels are associated with a lower risk of developing CVDs, while a higher testosterone/estradiol ratio is associated with a higher risk [324]. Moreover, the prognosis for women with heart failure is better than for men, implying a role of estrogens in heart regeneration [143]. Although some clinical studies in post-menopausal women demonstrate no impact of estrogen therapy on heart failure incidence [99, 177], trials in recently post-menopausal women do report a significantly reduced risk of heart failure and mortality [254], which led to the “timing hypothesis” [120]. Moreover, hormone therapy is associated with improved survival upon heart failure [173], and in particular after MI [262]. However, in a cohort of women with atrial fibrillation, hormone therapy is not associated with altered mortality [11], pointing to a multifactorial situation in vivo and the still-existing knowledge gap on underlying mechanisms.
Studies in ovariectomized female mice after experimentally induced MI demonstrate that hormone replacement attenuates cardiomyocyte apoptosis and reduces infarct size via activation of PI3K and AKT mediated by ERα [223], while other studies imply the induction of these signaling pathways by ERβ [306]. For example, cardiac function after MI is impaired in ERβ deficient mice, exhibiting prolonged ventricular repolarization and decreased automaticity compared to control mice after MI [155]. Interestingly, this effect seems to be gender-specific [306]. Conversely, cardiac function and survival after MI are improved in female and male mice, overexpressing ERβ in cardiomyocytes [255]. This effect is attributed to attenuated cardiac fibrosis but also to a more stable SERCA-2a expression and, thus, advanced calcium handling. Moreover, estrogen was recently shown to attenuate cardiomyocyte apoptosis after ischemia and reperfusion by a mechanism relying on 5-HT2BR expression, which is inhibited by glucocorticoids [68]. Finally, an estrogen-dependent modulation of the inflammatory response to cardiac injury is suggested to underlie an acceleration of heart regeneration by estrogen in zebrafish [318].
Even if preclinical studies imply a role of female sex hormones in heart regeneration, it must be borne in mind that the situation in patients is more complex. Differences in clinical outcomes between men and women are not necessarily due to hormonally altered regeneration of the myocardium but might also reflect differences in lifestyle or other confounding factors. Notably, similar to estrogen in women, low testosterone levels in men are associated with a higher incidence of CVDs and a worse prognosis [153]. Yet, preclinical and clinical studies on the effects of testosterone replacement therapy yield conflicting results, as comprehensively presented by Pongkan et al. [228]. Generally, hormones have various systemic effects, which is why it cannot be stated with certainty that functional improvement in clinical trials is solely due to hormone-induced tissue regeneration. Further research will be needed to decipher the implications of testosterone and other hormones in male and female cardiac health and regeneration.
Outlook
In December 2022, the Food and Drug Administration (FDA) announced that prior animal testing would no longer be required for human trials [302], reinforcing the role of potential substitutes, such as human PSC-derived cells, for large-scale research. In cardiology, hPSC-CMs have shown promise for drug and toxicity screening [160]. However, for reliable drug screening and more accurate disease modeling, it is crucial to overcome the immaturity of these cells. As discussed in this review, endocrine signaling may provide an underappreciated contribution to this goal. Importantly, beneficial effects on cardiomyocyte maturation are maximal when multiple hormones or hormone derivatives are applied, suggesting that future research should focus on such multifactorial approaches.
For therapeutic approaches, on the other hand, cardiomyocyte maturation might not be desirable as it has been shown that terminal maturation poses a barrier to regeneration. Intriguingly, however, the same hormones that have been demonstrated to promote the aspects of cardiomyocyte maturation have also been reported to have beneficial effects on cardiac regeneration. There are numerous promising preclinical trials of hormone-based strategies for cardiac regeneration, reporting beneficial effects on different aspects of ischemia–reperfusion injury (Fig. 3). Still, treatment with hormones and hormone substitutes has not yet found its way into clinical routine. Hormones are pleiotropic effectors, and their actions are highly dependent on a number of factors, including not only the developmental stage and gender but also levels of other hormones as well as tissue-specific physiological and pathophysiological conditions. Because of this, their widespread application for regeneration therapies seems to demand further extensive examination of the complex interactions within the endocrine system.

Consequences of ischemia–reperfusion and mechanisms underlying the beneficial effects of hormonal agents on these aspects
Data availability
No original datasets were generated for this review. All data supporting the information given here can be found in the references cited within the paper.
Abbreviations
- 5-HT2B :
-
5-Hydroxytryptamine receptor 2B
- ACTH:
-
Adrenocorticotropic hormone
- ACTN2:
-
α-Actinin 2
- ADP:
-
Adenosine diphosphate
- AKT:
-
Protein kinase B
- AMPK:
-
AMP-activated kinase
- ANG II:
-
Angiotensin 2
- ANP:
-
Atrial natriuretic peptide
- AR:
-
Androgen receptor
- ATP:
-
Adenosine triphosphate
- Atp2a2:
-
ATPase sarcoplasmic/endoplasmic reticulum Ca2 + transporting 2
- BNP:
-
Brain natriuretic peptide
- cAMP:
-
3′,5′-Cyclic adenosine monophosphate
- cGMP:
-
Cyclic guanosine monophosphate
- CH:
-
Corticosteroid hormone
- COX8:
-
Cytochrome c oxidase subunit 8
- cTnI:
-
Cardiac troponin I
- CVD:
-
Cardiovascular disease
- DIO1/2/3:
-
Deiodinases type I, II, and III
- DRP1:
-
Dynamin-related protein
- ER:
-
Estrogen receptor
- ERE:
-
Estrogen response element
- ERK:
-
Extracellular signal-regulated kinase
- ERR:
-
Estrogen-related receptor
- GATA4:
-
GATA-binding protein 4
- Gh:
-
Growth hormone
- GPER:
-
G-protein-coupled estrogen receptor
- GR:
-
Glucocorticoid receptor
- GRE:
-
Glucocorticoid response element
- Hcn2/4:
-
Hyperpolarization-activated cyclic nucleotide-gated ion channel 2 and 4
- HIF-1α:
-
Hypoxia-inducible factor 1α
- HK:
-
Hexokinase
- hPSC-CMs:
-
Human pluripotent stem cell-derived cardiomyocytes
- Igf1/2:
-
Insulin-like growth factor 1 and 2
- Igfbp:
-
Igf-binding protein
- IP3 :
-
Inositol 1,4,5-trisphosphate
- iPSCs:
-
Induced pluripotent stem cells
- JNK:
-
C-Jun N-terminal kinase
- MAPK:
-
Mitogen-activated protein kinases
- MEF2C:
-
Myocyte enhancer factor 2C
- MFN1/2:
-
Mitofusin 1 and 2
- MHC:
-
Myosin heavy chain
- MI:
-
Myocardial infarction
- mPR:
-
Membrane-bound progesterone receptor
- mTOR:
-
Mammalian target of rapamycin
- MYL:
-
Myosin light chain
- NCX:
-
Na+-Ca2+ exchanger
- Nkx2.5:
-
NK2 Homeobox 5
- NPR:
-
Natriuretic peptide receptors
- PGC1α:
-
Peroxisome proliferator-activated receptor γ coactivator 1α
- PI3K:
-
Phosphatidylinositol 3-kinase
- PKA:
-
Protein kinase A
- PKC:
-
Protein kinase C
- PKG:
-
Protein kinase G
- Plb:
-
Phospholamban
- PPAR:
-
Peroxisome proliferator-activated receptor
- RFP20/40:
-
Ring finger protein 20 and 40
- ROS:
-
Reactive oxygen species
- rT3:
-
3,3′,5′-L-triiodothyronine
- Ryr2:
-
Ryanodine receptor 2
- SERCA:
-
Sarcoplasmic/endoplasmic reticulum calcium ATPase
- SF3B2:
-
Splicing factor 3b Subunit 2
- SH:
-
Sex hormone
- Slc8a1 :
-
Solute carrier family 8 member A1
- ssTnI:
-
Slow skeletal troponin I
- T2:
-
3,3' Diiodo L thyronine
- T3:
-
Triiodothyronine
- T4:
-
Thyroxine
- TFAM:
-
Transcription factor A, mitochondrial
- TH:
-
Thyroid hormone
- TRE:
-
Thyroid response element
- TRα/β:
-
Thyroid receptors α and β
- TSH:
-
Thyroid-stimulating hormone
- YAP1:
-
Yes-associated protein-1
References
Abdul Kadir L, Stacey M, Barrett-Jolley R (2018) Emerging roles of the membrane potential: action beyond the action potential. Front Physiol. https://doi.org/10.3389/fphys.2018.01661
Agarkova I, Auerbach D, Ehler E, Perriard J-C (2000) A novel marker for vertebrate embryonic heart, the eh-myomesin isoform*. J Biol Chem 275:10256–10264. https://doi.org/10.1074/jbc.275.14.10256
Al Madhoun AS, Voronova A, Ryan T, Zakariyah A, McIntire C, Gibson L, Shelton M, Ruel M, Skerjanc IS (2013) Testosterone enhances cardiomyogenesis in stem cells and recruits the androgen receptor to the MEF2C and HCN4 genes. J Mol Cell Cardiol 60:164–171. https://doi.org/10.1016/j.yjmcc.2013.04.003
Alaynick WA, Kondo RP, Xie W, He W, Dufour CR, Downes M, Jonker JW, Giles W, Naviaux RK, Giguère V, Evans RM (2007) ERRγ directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab 6:13–24. https://doi.org/10.1016/j.cmet.2007.06.007
Alkass K, Panula J, Westman M, Wu T-D, Guerquin-Kern J-L, Bergmann O (2015) No evidence for cardiomyocyte number expansion in preadolescent mice. Cell 163:1026–1036. https://doi.org/10.1016/j.cell.2015.10.035
Almeida HV, Tenreiro MF, Louro AF, Abecasis B, Santinha D, Calmeiro T, Fortunato E, Ferreira L, Alves PM, Serra M (2021) Human extracellular-matrix functionalization of 3D hiPSC-based cardiac tissues improves cardiomyocyte maturation. ACS Appl Bio Mater 4:1888–1899. https://doi.org/10.1021/acsabm.0c01490
Almeida NAS, Cordeiro A, Machado DS, Souza LL, Ortiga-Carvalho TM, Campos-de-Carvalho AC, Wondisford FE, Pazos-Moura CC (2009) Connexin40 messenger ribonucleic acid is positively regulated by thyroid hormone (TH) acting in cardiac atria via the TH receptor. Endocrinology 150:546–554. https://doi.org/10.1210/en.2008-0451
Altamirano F, Oyarce C, Silva P, Toyos M, Wilson C, Lavandero S, Uhlén P, Estrada M (2009) Testosterone induces cardiomyocyte hypertrophy through mammalian target of rapamycin complex 1 pathway. J Endocrinol 202:299–307. https://doi.org/10.1677/JOE-09-0044
Anversa P (2005) Aging and Longevity. Circ Res 97:411–414. https://doi.org/10.1161/01.RES.0000182212.09147.56
Anyetei-Anum CS, Roggero VR, Allison LA (2018) Thyroid hormone receptor localization in target tissues. J Endocrinol 237:R19–R34. https://doi.org/10.1530/JOE-17-0708
Apostolakis S, Sullivan RM, Olshansky B, Lip GYH (2014) Hormone replacement therapy and adverse outcomes in women with atrial fibrillation: an analysis from the atrial fibrillation follow-up investigation of rhythm management trial. Stroke 45:3076–3079. https://doi.org/10.1161/STROKEAHA.114.006668
Ayrout M, Simon V, Bernard V, Binart N, Cohen-Tannoudji J, Lombès M, Chauvin S (2017) A novel non genomic glucocorticoid signaling mediated by a membrane palmitoylated glucocorticoid receptor cross talks with GnRH in gonadotrope cells. Sci Rep 7:1537. https://doi.org/10.1038/s41598-017-01777-2
Bačová BS, Vinczenzová C, Žurmanová J, Kašparová D, Knezl V, Beňová TE, Pavelka S, Soukup T, Tribulová N (2017) Altered thyroid status affects myocardial expression of connexin-43 and susceptibility of rat heart to malignant arrhythmias that can be partially normalized by red palm oil intake. Histochem Cell Biol 147:63–73. https://doi.org/10.1007/s00418-016-1488-6
Báez-Díaz C, Blanco-Blázquez V, Sánchez-Margallo F-M, Bayes-Genis A, González I, Abad A, Steendam R, Franssen O, Palacios I, Sánchez B, Gálvez-Montón C, Crisóstomo V (2020) Microencapsulated Insulin-Like Growth Factor-1 therapy improves cardiac function and reduces fibrosis in a porcine acute myocardial infarction model. Sci Rep 10:7166. https://doi.org/10.1038/s41598-020-64097-y
Bal MP, de Vries WB, Steendijk P, Homoet-van der Kraak P, van der Leij FR, Baan J, van Oosterhout MFM, van Bel F (2009) Histopathological changes of the heart after neonatal dexamethasone treatment: studies in 4-, 8-, and 50-week-old rats. Pediatr Res 66:74–79. https://doi.org/10.1203/PDR.0b013e3181a283a0
Bedada FB, Chan SS-K, Metzger SK, Zhang L, Zhang J, Garry DJ, Kamp TJ, Kyba M, Metzger JM (2014) Acquisition of a quantitative, stoichiometrically conserved ratiometric marker of maturation status in stem cell-derived cardiac myocytes. Stem Cell Reports 3:594–605. https://doi.org/10.1016/j.stemcr.2014.07.012
Beesley RD, Palmer BM, Casson PR, Toth MJ (2013) Effects of testosterone on cardiomyocyte calcium homeostasis and contractile function in female rats. Exp Physiol 98:161–171. https://doi.org/10.1113/expphysiol.2012.067009
Belakavadi M, Saunders J, Weisleder N, Raghava PS, Fondell JD (2010) Repression of cardiac phospholamban gene expression is mediated by thyroid hormone receptor-{alpha}1 and involves targeted covalent histone modifications. Endocrinology 151:2946–2956. https://doi.org/10.1210/en.2009-1241
van den Berg CW, Okawa S, de Sousa C, Lopes SM, van Iperen L, Passier R, Braam SR, Tertoolen LG, del Sol A, Davis RP, Mummery CL (2015) Transcriptome of human foetal heart compared with cardiomyocytes from pluripotent stem cells. Development 142:3231–3238. https://doi.org/10.1242/dev.123810
Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisén J (2009) Evidence for cardiomyocyte renewal in humans. Science 324:98–102. https://doi.org/10.1126/science.1164680
Bers DM (2002) Cardiac excitation–contraction coupling. Nature 415:198–205. https://doi.org/10.1038/415198a
Bersell K, Arab S, Haring B, Kühn B (2009) Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138:257–270. https://doi.org/10.1016/j.cell.2009.04.060
Bi W, Jia J, Pang R, Nie C, Han J, Ding Z, Liu B, Sheng R, Xu J, Zhang J (2019) Thyroid hormone postconditioning protects hearts from ischemia/reperfusion through reinforcing mitophagy. Biomed Pharmacother 118:109220. https://doi.org/10.1016/j.biopha.2019.109220
Bianco AC, Dumitrescu A, Gereben B, Ribeiro MO, Fonseca TL, Fernandes GW, Bocco BMLC (2019) Paradigms of dynamic control of thyroid hormone signaling. Endocr Rev 40:1000–1047. https://doi.org/10.1210/er.2018-00275
Binu AJ, Cherian KE, Kapoor N, Chacko ST, George O, Paul TV (2017) The heart of the matter: cardiac manifestations of endocrine disease. Indian J Endocrinol Metab 21:919–925. https://doi.org/10.4103/ijem.IJEM_212_17
Bird SD, Doevendans PA, van Rooijen MA, Brutel de la Riviere A, Hassink RJ, Passier R, Mummery CL (2003) The human adult cardiomyocyte phenotype. Cardiovasc Res 58:423–434. https://doi.org/10.1016/s0008-6363(03)00253-0
Blange I, Drvota V, Yen PM, Sylven C (1997) Species differences in cardiac thyroid hormone receptor isoforms protein abundance. Biol Pharm Bull 20:1123–1126. https://doi.org/10.1248/bpb.20.1123
de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H (1981) A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci 28:89–94. https://doi.org/10.1016/0024-3205(81)90370-2
Bots SH, Peters SAE, Woodward M (2017) Sex differences in coronary heart disease and stroke mortality: a global assessment of the effect of ageing between 1980 and 2010. BMJ Glob Health 2:e000298. https://doi.org/10.1136/bmjgh-2017-000298
Brodsky VY, Arefyeva AM, Gvasava IG, Sarkisov DS, Panova NW (1994) Polyploidy in cardiac myocytes of normal and hypertrophic human hearts; range of values. Vichows Archiv A Pathol Anat. https://doi.org/10.1007/BF00190566
Brüel A, Oxlund H, Nyengaard JR (2005) The total length of myocytes and capillaries, and total number of myocyte nuclei in the rat heart are time-dependently increased by growth hormone. Growth Hormon IGF Res 15:256–264. https://doi.org/10.1016/j.ghir.2005.04.003
Bruneau BG, de Bold AJ (1994) Selective changes in natriuretic peptide and early response gene expression in isolated rat atria following stimulation by stretch or endothelin-1. Cardiovasc Res 28:1519–1525. https://doi.org/10.1093/cvr/28.10.1519
Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM, Shoghi K, Welch MJ, Kelly DP (2007) Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest 117:3930–3939. https://doi.org/10.1172/JCI32578
Burridge PW, Keller G, Gold JD, Wu JC (2012) Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell 10:16–28. https://doi.org/10.1016/j.stem.2011.12.013
Bush IE (1953) Species differences in adrenocortical secretion. J Endocrinol 9:95-NP. https://doi.org/10.1677/joe.0.0090095
Cai W, Zhang J, de Lange WJ, Gregorich ZR, Karp H, Farrell ET, Mitchell SD, Tucholski T, Lin Z, Biermann M, McIlwain SJ, Ralphe JC, Kamp TJ, Ge Y (2019) An unbiased proteomics method to assess the maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Res 125:936–953. https://doi.org/10.1161/CIRCRESAHA.119.315305
Calmettes G, John SA, Weiss JN, Ribalet B (2013) Hexokinase–mitochondrial interactions regulate glucose metabolism differentially in adult and neonatal cardiac myocytes. J Gen Physiol 142:425–436. https://doi.org/10.1085/jgp.201310968
Caplice NM, DeVoe MC, Choi J, Dahly D, Murphy T, Spitzer E, Van Geuns R, Maher MM, Tuite D, Kerins DM, Ali MT, Kalyar I, Fahy EF, Khider W, Kelly P, Kearney PP, Curtin RJ, O’Shea C, Vaughan CJ, Eustace JA, McFadden EP (2018) Randomized placebo controlled trial evaluating the safety and efficacy of single low-dose intracoronary insulin-like growth factor following percutaneous coronary intervention in acute myocardial infarction (RESUS-AMI). Am Heart J 200:110–117. https://doi.org/10.1016/j.ahj.2018.03.018
Chapman K, Holmes M, Seckl J (2013) 11β-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev 93:1139–1206. https://doi.org/10.1152/physrev.00020.2012
Chattergoon NN (2019) Thyroid hormone signaling and consequences for cardiac development. J Endocrinol 242:T145–T160. https://doi.org/10.1530/JOE-18-0704
Chattergoon NN, Giraud GD, Louey S, Stork P, Fowden AL, Thornburg KL (2012) Thyroid hormone drives fetal cardiomyocyte maturation. FASEB J 26:397–408. https://doi.org/10.1096/fj.10-179895
Chattergoon NN, Louey S, Stork PJ, Giraud GD, Thornburg KL (2014) Unexpected maturation of PI3K and MAPK-ERK signaling in fetal ovine cardiomyocytes. Am J Physiol Heart Circ Physiol 307:H1216-1225. https://doi.org/10.1152/ajpheart.00833.2013
Chen Y, Lüttmann FF, Schoger E, Schöler HR, Zelarayán LC, Kim K-P, Haigh JJ, Kim J, Braun T (2021) Reversible reprogramming of cardiomyocytes to a fetal state drives heart regeneration in mice. Science 373:1537–1540. https://doi.org/10.1126/science.abg5159
Chen Y-F, Kobayashi S, Chen J, Redetzke RA, Said S, Liang Q, Gerdes AM (2008) Short term triiodo-L-thyronine treatment inhibits cardiac myocyte apoptosis in border area after myocardial infarction in rats. J Mol Cell Cardiol 44:180–187. https://doi.org/10.1016/j.yjmcc.2007.09.009
Chiba A, Watanabe-Takano H, Miyazaki T, Mochizuki N (2018) Cardiomyokines from the heart. Cell Mol Life Sci 75:1349–1362. https://doi.org/10.1007/s00018-017-2723-6
Chiba A, Watanabe-Takano H, Terai K, Fukui H, Miyazaki T, Uemura M, Hashimoto H, Hibi M, Fukuhara S, Mochizuki N (2016) Osteocrin, a peptide secreted from the heart and other tissues, contributes to cranial osteogenesis and chondrogenesis in zebrafish. Development. https://doi.org/10.1242/dev.143354
Chinkers M, Garbers DL, Chang MS, Lowe DG, Chin HM, Goeddel DV, Schulz S (1989) A membrane form of guanylate cyclase is an atrial natriuretic peptide receptor. Nature 338:78–83. https://doi.org/10.1038/338078a0
Chizzonite RA, Zak R (1984) Regulation of myosin isoenzyme composition in fetal and neonatal rat ventricle by endogenous thyroid hormones. J Biol Chem 259:12628–12632. https://doi.org/10.1016/S0021-9258(18)90792-1
Chong JJH, Yang X, Don CW, Minami E, Liu Y-W, Weyers JJ, Mahoney WM, Van Biber B, Cook SM, Palpant NJ, Gantz JA, Fugate JA, Muskheli V, Gough GM, Vogel KW, Astley CA, Hotchkiss CE, Baldessari A, Pabon L, Reinecke H, Gill EA, Nelson V, Kiem H-P, Laflamme MA, Murry CE (2014) Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 510:273–277. https://doi.org/10.1038/nature13233
Chu SH, Goldspink P, Kowalski J, Beck J, Schwertz DW (2006) Effect of estrogen on calcium-handling proteins, beta-adrenergic receptors, and function in rat heart. Life Sci 79:1257–1267. https://doi.org/10.1016/j.lfs.2006.03.037
Chu SH, Sutherland K, Beck J, Kowalski J, Goldspink P, Schwertz D (2005) Sex differences in expression of calcium-handling proteins and beta-adrenergic receptors in rat heart ventricle. Life Sci 76:2735–2749. https://doi.org/10.1016/j.lfs.2004.12.013
Cordeiro JM, Nesterenko VV, Sicouri S, Goodrow RJ, Treat JA, Desai M, Wu Y, Doss MX, Antzelevitch C, Di Diego JM (2013) Identification and characterization of a transient outward K+ current in human induced pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol 60:36–46. https://doi.org/10.1016/j.yjmcc.2013.03.014
Correia C, Koshkin A, Duarte P, Hu D, Teixeira A, Domian I, Serra M, Alves PM (2017) Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci Rep 7:8590. https://doi.org/10.1038/s41598-017-08713-4
Craelius W, Green WL, Harris DR (1990) Acute effects of thyroid hormone on sodium currents in neonatal myocytes. Biosci Rep 10:309–315. https://doi.org/10.1007/BF01117247
Crescioli C (2021) The role of estrogens and vitamin D in cardiomyocyte protection: a female perspective. Biomolecules 11:1815. https://doi.org/10.3390/biom11121815
Cruz-Topete D, Oakley RH, Cidlowski JA (2020) Glucocorticoid signaling and the aging heart. Front Endocrinol (Lausanne) 11:347. https://doi.org/10.3389/fendo.2020.00347
Curl CL, Wendt IR, Canny BJ, Kotsanas G (2003) Effects of ovariectomy and 17 beta-oestradiol replacement on [Ca2+]i in female rat cardiac myocytes. Clin Exp Pharmacol Physiol 30:489–494. https://doi.org/10.1046/j.1440-1681.2003.03864.x
Dahms NM, Lobel P, Kornfeld S (1989) Mannose 6-phosphate receptors and lysosomal enzyme targeting. J Biol Chem 264:12115–12118. https://doi.org/10.1016/S0021-9258(18)63825-6
Darras VM, Kotanen SP, Geris KL, Berghman LR, Kühn ER (1996) Plasma thyroid hormone levels and iodothyronine deiodinase activity following an acute glucocorticoid challenge in embryonic compared with posthatch chickens. Gen Comp Endocrinol 104:203–212. https://doi.org/10.1006/gcen.1996.0163
Daughaday WH, Parker KA, Borowsky S, Trivedi B, Kapadia M (1982) Measurement of somatomedin-related peptides in fetal, neonatal, and maternal rat serum by insulin-like growth factor (IGF) I radioimmunoassay, IGF-II radioreceptor assay (RRA), and multiplication-stimulating activity RRA after acid-ethanol extraction*. Endocrinology 110:575–581. https://doi.org/10.1210/endo-110-2-575
Davis ME, Hsieh PCH, Takahashi T, Song Q, Zhang S, Kamm RD, Grodzinsky AJ, Anversa P, Lee RT (2006) Local myocardial insulin-like growth factor 1 (IGF-1) delivery with biotinylated peptide nanofibers improves cell therapy for myocardial infarction. Proc Natl Acad Sci U S A 103:8155–8160. https://doi.org/10.1073/pnas.0602877103
Davis PJ, Lin H-Y, Mousa SA, Luidens MK, Hercbergs AA, Wehling M, Davis FB (2011) Overlapping nongenomic and genomic actions of thyroid hormone and steroids. Steroids 76:829–833. https://doi.org/10.1016/j.steroids.2011.02.012
De Bosscher K, Beck IM, Dejager L, Bougarne N, Gaigneaux A, Chateauvieux S, Ratman D, Bracke M, Tavernier J, Vanden Berghe W, Libert C, Diederich M, Haegeman G (2014) Selective modulation of the glucocorticoid receptor can distinguish between transrepression of NF-κB and AP-1. Cell Mol Life Sci 71:143–163. https://doi.org/10.1007/s00018-013-1367-4
Deftos LJ, Burton DW, Brandt DW (1993) Parathyroid hormone-like protein is a secretory product of atrial myocytes. J Clin Invest 92:727–735
DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, Hinson JT, Homsy J, Gray J, Pu W, Bruneau BG, Seidman JG, Seidman CE (2016) Single-cell resolution of temporal gene expression during heart development. Dev Cell 39:480–490. https://doi.org/10.1016/j.devcel.2016.10.001
Delaughter MC, Taffet GE, Fiorotto ML, Entman ML, Schwartz RJ (1999) Local insulin-like growth factor I expression induces physiologic, then pathologic, cardiac hypertrophy in transgenic mice. FASEB J 13:1923–1929. https://doi.org/10.1096/fasebj.13.14.1923
D’Ercole AJ, Stiles AD, Underwood LE (1984) Tissue concentrations of somatomedin C: further evidence for multiple sites of synthesis and paracrine or autocrine mechanisms of action. Proc Natl Acad Sci U S A 81:935–939. https://doi.org/10.1073/pnas.81.3.935
Dhaibar HA, Carroll NG, Amatya S, Kamberov L, Khanna P, Orr AW, Bailey SR, Oakley RH, Cidlowski JA, Cruz-Topete D (2021) Glucocorticoid inhibition of estrogen regulation of the serotonin receptor 2B in cardiomyocytes exacerbates cell death in hypoxia/reoxygenation injury. J Am Heart Assoc 10:e015868. https://doi.org/10.1161/JAHA.120.015868
Donath MY, Zapf J, Eppenberger-Eberhardt M, Froesch ER, Eppenberger HM (1994) Insulin-like growth factor I stimulates myofibril development and decreases smooth muscle alpha-actin of adult cardiomyocytes. Proc Natl Acad Sci USA 91:1686–1690. https://doi.org/10.1073/pnas.91.5.1686
Dudley SC, Baumgarten CM (1993) Bursting of cardiac sodium channels after acute exposure to 3,5,3’-triiodo-L-thyronine. Circ Res 73:301–313. https://doi.org/10.1161/01.RES.73.2.301
Ellis MJ, Leav BA, Yang Z, Rasmussen A, Pearce A, Zweibel JA, Lippman ME, Cullen KJ (1996) Affinity for the insulin-like growth factor-II (IGF-II) receptor inhibits autocrine IGF-II activity in MCF-7 breast cancer cells. Mol Endocrinol 10:286–297. https://doi.org/10.1210/mend.10.3.8833657
England J, Loughna S (2013) Heavy and light roles: myosin in the morphogenesis of the heart. Cell Mol Life Sci 70:1221–1239. https://doi.org/10.1007/s00018-012-1131-1
Eshak ES, Maruyama K, Iso H, Tamakoshi A (2019) The Prospective association between plasma concentrations of cellular growth factors and risk of heart failure mortality in Japanese population. J Epidemiol 29:104–109. https://doi.org/10.2188/jea.JE20170123
Fahmi AI, Forhead AJ, Fowden AL, Vandenberg JI (2004) Cortisol influences the ontogeny of both alpha- and beta-subunits of the cardiac sodium channel in fetal sheep. J Endocrinol 180:449–455. https://doi.org/10.1677/joe.0.1800449
Fisher ffolliott M, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, Spiegelman BM (2012) FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev 26:271–281. https://doi.org/10.1101/gad.177857.111
Foradori CD, Weiser MJ, Handa RJ (2008) Non-genomic actions of androgens. Front Neuroendocrinol 29:169–181. https://doi.org/10.1016/j.yfrne.2007.10.005
Forini F, Lionetti V, Ardehali H, Pucci A, Cecchetti F, Ghanefar M, Nicolini G, Ichikawa Y, Nannipieri M, Recchia FA, Iervasi G (2011) Early long-term L-T3 replacement rescues mitochondria and prevents ischemic cardiac remodelling in rats. J Cell Mol Med 15:514–524. https://doi.org/10.1111/j.1582-4934.2010.01014.x
Fowden AL, Li J, Forhead AJ (1998) Glucocorticoids and the preparation for life after birth: are there long-term consequences of the life insurance? Proc Nutr Soc 57:113–122. https://doi.org/10.1079/pns19980017
Friedrichsen S, Christ S, Heuer H, Schäfer MKH, Mansouri A, Bauer K, Visser TJ (2003) Regulation of iodothyronine deiodinases in the Pax8−/− mouse model of congenital hypothyroidism. Endocrinology 144:777–784. https://doi.org/10.1210/en.2002-220715
Garbern JC, Helman A, Sereda R, Sarikhani M, Ahmed A, Escalante GO, Ogurlu R, Kim SL, Zimmerman JF, Cho A, MacQueen L, Bezzerides VJ, Parker KK, Melton DA, Lee RT (2020) Inhibition of mTOR signaling enhances maturation of cardiomyocytes derived from human-induced pluripotent stem cells via p53-induced quiescence. Circulation 141:285–300. https://doi.org/10.1161/CIRCULATIONAHA.119.044205
Garbern JC, Lee RT (2021) Mitochondria and metabolic transitions in cardiomyocytes: lessons from development for stem cell-derived cardiomyocytes. Stem Cell Res Ther 12:177. https://doi.org/10.1186/s13287-021-02252-6
Garcia RA, Roemmich JN, Claycombe KJ (2016) Evaluation of markers of beige adipocytes in white adipose tissue of the mouse. Nutr Metab (Lond) 13:24. https://doi.org/10.1186/s12986-016-0081-2
Gassanov N, Er F, Endres-Becker J, Wolny M, Schramm C, Hoppe UC (2009) Distinct regulation of cardiac I(f) current via thyroid receptors alpha1 and beta1. Pflugers Arch 458:1061–1068. https://doi.org/10.1007/s00424-009-0691-x
Gentillon C, Li D, Duan M, Yu W-M, Preininger MK, Jha R, Rampoldi A, Saraf A, Gibson GC, Qu C-K, Brown LA, Xu C (2019) Targeting HIF-1α in combination with PPARα activation and postnatal factors promotes the metabolic maturation of human induced pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol 132:120–135. https://doi.org/10.1016/j.yjmcc.2019.05.003
Gerdes AM, Kellerman SE, Moore JA, Muffly KE, Clark LC, Reaves PY, Malec KB, McKeown PP, Schocken DD (1992) Structural remodeling of cardiac myocytes in patients with ischemic cardiomyopathy. Circulation 86:426–430. https://doi.org/10.1161/01.cir.86.2.426
Giguére V (2002) To ERR in the estrogen pathway. Trends Endocrinol Metab 13:220–225. https://doi.org/10.1016/S1043-2760(02)00592-1
Giguère V (2008) Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev 29:677–696. https://doi.org/10.1210/er.2008-0017
Giguère V, Yang N, Segui P, Evans RM (1988) Identification of a new class of steroid hormone receptors. Nature 331:91–94. https://doi.org/10.1038/331091a0
Gloss B, Trost S, Bluhm W, Swanson E, Clark R, Winkfein R, Janzen K, Giles W, Chassande O, Samarut J, Dillmann W (2001) Cardiac ion channel expression and contractile function in mice with deletion of thyroid hormone receptor alpha or beta. Endocrinology 142:544–550. https://doi.org/10.1210/endo.142.2.7935
Gluckman PD, Butler JH (1983) Parturition-related changes in insulin-like growth factors-I and -II in the perinatal lamb. J Endocrinol 99:223–232. https://doi.org/10.1677/joe.0.0990223
Golden KL, Marsh JD, Jiang Y (2004) Testosterone regulates mRNA levels of calcium regulatory proteins in cardiac myocytes. Horm Metab Res 36:197–202. https://doi.org/10.1055/s-2004-814445
Golden KL, Marsh JD, Jiang Y, Moulden J (2005) Acute actions of testosterone on contractile function of isolated rat ventricular myocytes. Eur J Endocrinol 152:479–483. https://doi.org/10.1530/eje.1.01845
Goldenthal MJ, Weiss HR, Marín-García J (2004) Bioenergetic remodeling of heart mitochondria by thyroid hormone. Mol Cell Biochem 265:97–106. https://doi.org/10.1023/b:mcbi.0000044321.17680.a2
Goldman-Johnson DR, de Kretser DM, Morrison JR (2008) Evidence that androgens regulate early developmental events, prior to sexual differentiation. Endocrinology 149:5–14. https://doi.org/10.1210/en.2007-1123
Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW (2015) Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350:aad2459. https://doi.org/10.1126/science.aad2459
Gong Y, Yang J, Liu Q, Cai J, Zheng Y, Zhang Y, Yu D, Liu H, Zhang Z (2019) IGF1 knockdown hinders myocardial development through energy metabolism dysfunction caused by ROS-dependent FOXO activation in the chicken heart. Oxid Med Cell Longev 2019:7838754. https://doi.org/10.1155/2019/7838754
Gonzalez-Reyes A, Menaouar A, Yip D, Danalache B, Plante E, Noiseux N, Gutkowska J, Jankowski M (2015) Molecular mechanisms underlying oxytocin-induced cardiomyocyte protection from simulated ischemia-reperfusion. Mol Cell Endocrinol 412:170–181. https://doi.org/10.1016/j.mce.2015.04.028
Gower WR, McCuen RW, Arimura A, Coy DA, Dietz JR, Landon CS, Schubert ML (2003) Reciprocal paracrine pathways link atrial natriuretic peptide and somatostatin secretion in the antrum of the stomach. Regul Pept 110:101–106. https://doi.org/10.1016/s0167-0115(02)00206-9
Grady D, Herrington D, Bittner V, Blumenthal R, Davidson M, Hlatky M, Hsia J, Hulley S, Herd A, Khan S, Newby LK, Waters D, Vittinghoff E, Wenger N, HERS Research Group (2002) Cardiovascular disease outcomes during 6.8 years of hormone therapy: heart and Estrogen/progestin Replacement Study follow-up (HERS II). JAMA 288:49–57. https://doi.org/10.1001/jama.288.1.49
Graham N, Huang GN (2021) Endocrine influence on cardiac metabolism in development and regeneration. Endocrinology 162:bqab081. https://doi.org/10.1210/endocr/bqab081
Grant AO (2009) Cardiac ion channels. Circ Arrhyth Electrophysiol 2:185–194. https://doi.org/10.1161/CIRCEP.108.789081
Green CR, Severs NJ (1993) Distribution and role of gap junctions in normal myocardium and human ischaemic heart disease. Histochemistry 99:105–120. https://doi.org/10.1007/BF00571871
Grimm D, Cameron D, Griese DP, Riegger GAJ, Kromer EP (1998) Differential effects of growth hormone on cardiomyocyte and extracellular matrix protein remodeling following experimental myocardial infarction. Cardiovasc Res 40:297–306. https://doi.org/10.1016/S0008-6363(98)00181-3
Gunata M, Parlakpinar H (2021) A review of myocardial ischaemia/reperfusion injury: pathophysiology, experimental models, biomarkers, genetics and pharmacological treatment. Cell Biochem Funct 39:190–217. https://doi.org/10.1002/cbf.3587
Guo Y, Cao Y, Jardin BD, Sethi I, Ma Q, Moghadaszadeh B, Troiano EC, Mazumdar N, Trembley MA, Small EM, Yuan G-C, Beggs AH, Pu WT (2021) Sarcomeres regulate murine cardiomyocyte maturation through MRTF-SRF signaling. Proc Natl Acad Sci 118:e2008861118. https://doi.org/10.1073/pnas.2008861118
Guo Y, Jardin BD, Zhou P, Sethi I, Akerberg BN, Toepfer CN, Ai Y, Li Y, Ma Q, Guatimosim S, Hu Y, Varuzhanyan G, VanDusen NJ, Zhang D, Chan DC, Yuan G-C, Seidman CE, Seidman JG, Pu WT (2018) Hierarchical and stage-specific regulation of murine cardiomyocyte maturation by serum response factor. Nat Commun 9:3837. https://doi.org/10.1038/s41467-018-06347-2
Guo Y, Pu WT (2020) Cardiomyocyte maturation. Circ Res 126:1086–1106. https://doi.org/10.1161/CIRCRESAHA.119.315862
Gupta MP (2007) Factors controlling cardiac myosin-isoform shift during hypertrophy and heart failure. J Mol Cell Cardiol 43:388–403. https://doi.org/10.1016/j.yjmcc.2007.07.045
Gupta S, Mitra A (2021) Heal the heart through gut (hormone) ghrelin: a potential player to combat heart failure. Heart Fail Rev 26:417–435. https://doi.org/10.1007/s10741-020-10032-2
Gustafson TA, Markham BE, Morkin E (1986) Effects of thyroid hormone on alpha-actin and myosin heavy chain gene expression in cardiac and skeletal muscles of the rat: measurement of mRNA content using synthetic oligonucleotide probes. Circ Res 59:194–201. https://doi.org/10.1161/01.RES.59.2.194
Haddad F, Jiang W, Bodell PW, Qin AX, Baldwin KM (2010) Cardiac myosin heavy chain gene regulation by thyroid hormone involves altered histone modifications. Am J Physiol-Heart Circ Physiol 299:H1968–H1980. https://doi.org/10.1152/ajpheart.00644.2010
Halbach M, Krausgrill B, Hannes T, Wiedey M, Peinkofer G, Baumgartner S, Sahito RGA, Pfannkuche K, Pillekamp F, Reppel M, Müller-Ehmsen J, Hescheler J (2012) Time-course of the electrophysiological maturation and integration of transplanted cardiomyocytes. J Mol Cell Cardiol 53:401–408. https://doi.org/10.1016/j.yjmcc.2012.06.007
Hamilton MA, Stevenson LW, Fonarow GC, Steimle A, Goldhaber JI, Child JS, Chopra IJ, Moriguchi JD, Hage A (1998) Safety and hemodynamic effects of intravenous triiodothyronine in advanced congestive heart failure. Am J Cardiol 81:443–447. https://doi.org/10.1016/s0002-9149(97)00950-8
Hammes A, Andreassen TK, Spoelgen R, Raila J, Hubner N, Schulz H, Metzger J, Schweigert FJ, Luppa PB, Nykjaer A, Willnow TE (2005) Role of endocytosis in cellular uptake of sex steroids. Cell 122:751–762. https://doi.org/10.1016/j.cell.2005.06.032
Haufe V, Camacho JA, Dumaine R, Günther B, Bollensdorff C, von Banchet GS, Benndorf K, Zimmer T (2005) Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J Physiol 564:683–696. https://doi.org/10.1113/jphysiol.2004.079681
Heinen A, Nederlof R, Panjwani P, Spychala A, Tschaidse T, Reffelt H, Boy J, Raupach A, Gödecke S, Petzsch P, Köhrer K, Grandoch M, Petz A, Fischer JW, Alter C, Vasilevska J, Lang P, Gödecke A (2019) IGF1 Treatment improves cardiac remodeling after infarction by targeting myeloid cells. Mol Ther 27:46–58. https://doi.org/10.1016/j.ymthe.2018.10.020
Hellen N, Wheeler J, Pinto Ricardo C, Foldes G, Kodagoda T, Whiting G, Mioulane M, Terracciano C, Vauchez K, Harding S (2014) P343Effect of T3 on human induced pluripotent stem cell-derived cardiomyocyte maturation. Cardiovasc Res 103:S62. https://doi.org/10.1093/cvr/cvu091.29
Hiller-Sturmhöfel S, Bartke A (1998) The endocrine system. Alcohol Health Res World 22:153–164
Hirose K, Payumo AY, Cutie S, Hoang A, Zhang H, Guyot R, Lunn D, Bigley RB, Yu H, Wang J, Smith M, Gillett E, Muroy SE, Schmid T, Wilson E, Field KA, Reeder DM, Maden M, Yartsev MM, Wolfgang MJ, Grützner F, Scanlan TS, Szweda LI, Buffenstein R, Hu G, Flamant F, Olgin JE, Huang GN (2019) Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 364:184–188. https://doi.org/10.1126/science.aar2038
Hodis HN, Mack WJ (2022) Menopausal hormone replacement therapy and reduction of all-cause mortality and cardiovascular disease: it is about time and timing. Cancer J 28:208–223. https://doi.org/10.1097/PPO.0000000000000591
Horikoshi Y, Yan Y, Terashvili M, Wells C, Horikoshi H, Fujita S, Bosnjak ZJ, Bai X (2019) Fatty acid-treated induced pluripotent stem cell-derived human cardiomyocytes exhibit adult cardiomyocyte-like energy metabolism phenotypes. Cells 8:1095. https://doi.org/10.3390/cells8091095
Hu D, Linders A, Yamak A, Correia C, Kijlstra JD, Garakani A, Xiao L, Milan DJ, van der Meer P, Serra M, Alves PM, Domian IJ (2018) Metabolic maturation of human pluripotent stem cell derived cardiomyocytes by inhibition of HIF1α and LDHA. Circ Res 123:1066–1079. https://doi.org/10.1161/CIRCRESAHA.118.313249
Hu P, Liu J, Zhao J, Wilkins BJ, Lupino K, Wu H, Pei L (2018) Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev 32:1344–1357. https://doi.org/10.1101/gad.316802.118
Huang CY, Peres Moreno Maia-Joca R, Ong CS, Wilson I, DiSilvestre D, Tomaselli GF, Reich DH (2020) Enhancement of human iPSC-derived cardiomyocyte maturation by chemical conditioning in a 3D environment. J Mol Cell Cardiol 138:1–11. https://doi.org/10.1016/j.yjmcc.2019.10.001
Huang Y, Harrison MR, Osorio A, Kim J, Baugh A, Duan C, Sucov HM, Lien C-L (2013) Igf signaling is required for cardiomyocyte proliferation during zebrafish heart development and regeneration. PLoS ONE 8:e67266. https://doi.org/10.1371/journal.pone.0067266
Ibarra C, Estrada M, Carrasco L, Chiong M, Liberona JL, Cardenas C, Díaz-Araya G, Jaimovich E, Lavandero S (2004) Insulin-like growth factor-1 induces an inositol 1,4,5-trisphosphate-dependent increase in nuclear and cytosolic calcium in cultured rat cardiac myocytes*. J Biol Chem 279:7554–7565. https://doi.org/10.1074/jbc.M311604200
Inukai T, Takanashi K, Takebayashi K, Fujiwara Y, Tayama K, Takemura Y (1999) Thyroid hormone modulates insulin-like growth factor-I(IGF-I) and IGF-binding protein-3, without mediation by growth hormone, in patients with autoimmune thyroid diseases. Horm Metab Res 31:576–579. https://doi.org/10.1055/s-2007-978798
Ito H, Hiroe M, Hirata Y, Tsujino M, Adachi S, Shichiri M, Koike A, Nogami A, Marumo F (1993) Insulin-like growth factor-I induces hypertrophy with enhanced expression of muscle specific genes in cultured rat cardiomyocytes. Circulation 87:1715–1721. https://doi.org/10.1161/01.cir.87.5.1715
Ivashchenko CY, Pipes GC, Lozinskaya IM, Lin Z, Xiaoping X, Needle S, Grygielko ET, Hu E, Toomey JR, Lepore JJ, Willette RN (2013) Human-induced pluripotent stem cell-derived cardiomyocytes exhibit temporal changes in phenotype. Am J Physiol-Heart Circ Physiol 305:H913–H922. https://doi.org/10.1152/ajpheart.00819.2012
Ivy JR, Carter RN, Zhao J-F, Buckley C, Urquijo H, Rog-Zielinska EA, Panting E, Hrabalkova L, Nicholson C, Agnew EJ, Kemp MW, Morton NM, Stock SJ, Wyrwoll C, Ganley IG, Chapman KE (2021) Glucocorticoids regulate mitochondrial fatty acid oxidation in fetal cardiomyocytes. J Physiol 599:4901–4924. https://doi.org/10.1113/JP281860
Jackman C, Li H, Bursac N (2018) Long-term contractile activity and thyroid hormone supplementation produce engineered rat myocardium with adult-like structure and function. Acta Biomater 78:98–110. https://doi.org/10.1016/j.actbio.2018.08.003
Jankowski M, Hajjar F, Kawas SA, Mukaddam-Daher S, Hoffman G, McCann SM, Gutkowska J (1998) Rat heart: a site of oxytocin production and action. Proc Natl Acad Sci U S A 95:14558–14563. https://doi.org/10.1073/pnas.95.24.14558
Jeon YG, Lee JH, Ji Y, Sohn JH, Lee D, Kim DW, Yoon SG, Shin KC, Park J, Seong JK, Cho J-Y, Choe SS, Kim JB (2019) RNF20 functions as a transcriptional coactivator for PPARγ by promoting NCoR1 degradation in adipocytes. Diabetes 69:20–34. https://doi.org/10.2337/db19-0508
Jiang Y, Lian XL (2020) Heart regeneration with human pluripotent stem cells: prospects and challenges. Bioact Mater 5:74–81. https://doi.org/10.1016/j.bioactmat.2020.01.003
Johansson C, Vennström B, Thorén P (1998) Evidence that decreased heart rate in thyroid hormone receptor-alpha1-deficient mice is an intrinsic defect. Am J Physiol 275:R640-646. https://doi.org/10.1152/ajpregu.1998.275.2.R640
Jonker SS, Louey S (2016) Endocrine and other physiologic modulators of perinatal cardiomyocyte endowment. J Endocrinol 228:R1-18. https://doi.org/10.1530/JOE-15-0309
Jonker SS, Louey S, Roselli CE (2018) Cardiac myocyte proliferation and maturation near term is inhibited by early gestation maternal testosterone exposure. Am J Physiol-Heart Circ Physiol 315:H1393–H1401. https://doi.org/10.1152/ajpheart.00314.2018
Kajstura J, Cheng W, Reiss K, Anversa P (1994) The IGF-1-IGF-1 receptor system modulates myocyte proliferation but not myocyte cellular hypertrophy in vitro. Exp Cell Res 215:273–283. https://doi.org/10.1006/excr.1994.1343
Kalász J, Tóth EP, Bódi B, Fagyas M, Tóth A, Pal BH, Vári SG, Balog M, Blažetić S, Heffer M, Papp Z, Borbély A (2014) Single acute stress-induced progesterone and ovariectomy alter cardiomyocyte contractile function in female rats. Croat Med J 55:239–249. https://doi.org/10.3325/cmj.2014.55.239
Kamakura T, Makiyama T, Sasaki K, Yoshida Y, Wuriyanghai Y, Chen J, Hattori T, Ohno S, Kita T, Horie M, Yamanaka S, Kimura T (2013) Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ J 77:1307–1314. https://doi.org/10.1253/circj.cj-12-0987
Kanashiro-Takeuchi RM, Tziomalos K, Takeuchi LM, Treuer AV, Lamirault G, Dulce R, Hurtado M, Song Y, Block NL, Rick F, Klukovits A, Hu Q, Varga JL, Schally AV, Hare JM (2010) Cardioprotective effects of growth hormone-releasing hormone agonist after myocardial infarction. Proc Natl Acad Sci USA 107:2604–2609. https://doi.org/10.1073/pnas.0914138107
Kane LA, Youle RJ (2010) Mitochondrial fission and fusion and their roles in the heart. J Mol Med (Berl) 88:971–979. https://doi.org/10.1007/s00109-010-0674-6
Kannel WB (2002) The Framingham Study: historical insight on the impact of cardiovascular risk factors in men versus women. J Gend Specif Med 5:27–37
Karbassi E, Fenix A, Marchiano S, Muraoka N, Nakamura K, Yang X, Murry CE (2020) Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat Rev Cardiol 17:341–359. https://doi.org/10.1038/s41569-019-0331-x
van Kempen MJ, Fromaget C, Gros D, Moorman AF, Lamers WH (1991) Spatial distribution of connexin43, the major cardiac gap junction protein, in the developing and adult rat heart. Circ Res 68:1638–1651. https://doi.org/10.1161/01.res.68.6.1638
Kenessey A, Ojamaa K (2006) Thyroid hormone stimulates protein synthesis in the cardiomyocyte by activating the Akt-mTOR and p70S6K Pathways*. J Biol Chem 281:20666–20672. https://doi.org/10.1074/jbc.M512671200
Khait L, Birla RK (2008) Effect of thyroid hormone on the contractility of self-organized heart muscle. In Vitro Cell DevBiol-Animal 44:204–213. https://doi.org/10.1007/s11626-008-9094-0
Khan MA, Hashim MJ, Mustafa H, Baniyas MY, Al Suwaidi SKBM, AlKatheeri R, Alblooshi FMK, Almatrooshi MEAH, Alzaabi MEH, Al Darmaki RS, Lootah SNAH (2020) Global epidemiology of ischemic heart disease: results from the global burden of disease study. Cureus. 12:e9349. https://doi.org/10.7759/cureus.9349
Khosla S, Monroe DG (2018) Regulation of Bone Metabolism by Sex Steroids. Cold Spring Harb Perspect Med 8:a031211. https://doi.org/10.1101/cshperspect.a031211
Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, Kan NG, Forcales S, Puri PL, Leone TC, Marine JE, Calkins H, Kelly DP, Judge DP, Chen H-SV (2013) Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 494:105–110. https://doi.org/10.1038/nature11799
Kim HD, Kim DJ, Lee IJ, Rah BJ, Sawa Y, Schaper J (1992) Human fetal heart development after mid-term: morphometry and ultrastructural study. J Mol Cell Cardiol 24:949–965. https://doi.org/10.1016/0022-2828(92)91862-y
Kim MY, Eiby YA, Lumbers ER, Wright LL, Gibson KJ, Barnett AC, Lingwood BE (2014) Effects of glucocorticoid exposure on growth and structural maturation of the heart of the preterm piglet. PLoS ONE 9:e93407. https://doi.org/10.1371/journal.pone.0093407
Kloner RA, Carson C, Dobs A, Kopecky S, Mohler ER (2016) Testosterone and cardiovascular disease. J Am Coll Cardiol 67:545–557. https://doi.org/10.1016/j.jacc.2015.12.005
Knowlton AA, Lee AR (2012) Estrogen and the cardiovascular system. Pharmacol Ther 135:54–70. https://doi.org/10.1016/j.pharmthera.2012.03.007
Korte T, Fuchs M, Arkudas A, Geertz S, Meyer R, Gardiwal A, Klein G, Niehaus M, Krust A, Chambon P, Drexler H, Fink K, Grohé C (2005) Female mice lacking estrogen receptor beta display prolonged ventricular repolarization and reduced ventricular automaticity after myocardial infarction. Circulation 111:2282–2290. https://doi.org/10.1161/01.CIR.0000164262.08004.BB
Kravtsov GM, Kam KWL, Liu J, Wu S, Wong TM (2007) Altered Ca2+ handling by ryanodine receptor and Na+-Ca2+ exchange in the heart from ovariectomized rats: role of protein kinase A. Am J Physiol Cell Physiol 292:C1625–C1635. https://doi.org/10.1152/ajpcell.00368.2006
Krüger M, Sachse C, Zimmermann WH, Eschenhagen T, Klede S, Linke WA (2008) Thyroid hormone regulates developmental titin isoform transitions via the phosphatidylinositol-3-kinase/ AKT pathway. Circ Res 102:439–447. https://doi.org/10.1161/CIRCRESAHA.107.162719
Kubalak SW, Miller-Hance WC, O’Brien TX, Dyson E, Chien KR (1994) Chamber specification of atrial myosin light chain-2 expression precedes septation during murine cardiogenesis. J Biol Chem 269:16961–16970. https://doi.org/10.1016/S0021-9258(19)89483-8
Kühn B, del Monte F, Hajjar RJ, Chang Y-S, Lebeche D, Arab S, Keating MT (2007) Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med 13:962–969. https://doi.org/10.1038/nm1619
Kussauer S, David R, Lemcke H (2019) hiPSCs derived cardiac cells for drug and toxicity screening and disease modeling: what micro- electrode-array analyses can tell us. Cells 8:1331. https://doi.org/10.3390/cells8111331
La Mear NS, MacGilvray SS, Myers TF (1997) Dexamethasone-induced myocardial hypertrophy in neonatal rats. Biol Neonate 72:175–180. https://doi.org/10.1159/000244481
Laustsen PG, Russell SJ, Cui L, Entingh-Pearsall A, Holzenberger M, Liao R, Kahn CR (2007) Essential role of insulin and insulin-like growth factor 1 receptor signaling in cardiac development and function. Mol Cell Biol 27:1649–1664. https://doi.org/10.1128/MCB.01110-06
Lee WL, Chen JW, Ting CT, Lin SJ, Wang PH (1999) Changes of the insulin-like growth factor I system during acute myocardial infarction: implications on left ventricular remodeling. J Clin Endocrinol Metab 84:1575–1581. https://doi.org/10.1210/jcem.84.5.5676
Lee Y-K, Ng K-M, Chan Y-C, Lai W-H, Au K-W, Ho C-YJ, Wong L-Y, Lau C-P, Tse H-F, Siu C-W (2010) Triiodothyronine promotes cardiac differentiation and maturation of embryonic stem cells via the classical genomic pathway. Mol Endocrinol 24:1728–1736. https://doi.org/10.1210/me.2010-0032
Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP (2000) Peroxisome proliferator–activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest 106:847–856. https://doi.org/10.1172/JCI10268
Lewandowski J, Rozwadowska N, Kolanowski TJ, Malcher A, Zimna A, Rugowska A, Fiedorowicz K, Łabędź W, Kubaszewski Ł, Chojnacka K, Bednarek-Rajewska K, Majewski P, Kurpisz M (2018) The impact of in vitro cell culture duration on the maturation of human cardiomyocytes derived from induced pluripotent stem cells of myogenic origin. Cell Transplant 27:1047–1067. https://doi.org/10.1177/0963689718779346
LeWinter MM, Granzier H (2010) Cardiac titin—a multifunctional giant. Circulation 121:2137–2145. https://doi.org/10.1161/CIRCULATIONAHA.109.860171
Li F (1996) Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 28:1737–1746. https://doi.org/10.1006/jmcc.1996.0163
Li Q, Wu S, Li S-Y, Lopez FL, Du M, Kajstura J, Anversa P, Ren J (2007) Cardiac-specific overexpression of insulin-like growth factor 1 attenuates aging-associated cardiac diastolic contractile dysfunction and protein damage. Am J Physiol-Heart Circ Physiol 292:H1398–H1403. https://doi.org/10.1152/ajpheart.01036.2006
Lieu DK, Fu J-D, Chiamvimonvat N, Tung KC, McNerney GP, Huser T, Keller G, Kong C-W, Li RA (2013) Mechanism-based facilitated maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Arrhythm Electrophysiol 6:191–201. https://doi.org/10.1161/CIRCEP.111.973420
Lieu DK, Liu J, Siu C-W, McNerney GP, Tse H-F, Abu-Khalil A, Huser T, Li RA (2009) Absence of transverse tubules contributes to non-uniform Ca2+ wavefronts in mouse and human embryonic stem cell-derived cardiomyocytes. Stem Cells Dev 18:1493–1500. https://doi.org/10.1089/scd.2009.0052
Lindberg T, Wimo A, Elmståhl S, Qiu C, Bohman DM, Sanmartin Berglund J (2019) Prevalence and incidence of atrial fibrillation and other arrhythmias in the general older population: findings from the Swedish national study on aging and care. Gerontol Geriatr Med. https://doi.org/10.1177/2333721419859687
Lindenfeld J, Ghali JK, Krause-Steinrauf HJ, Khan S, Adams K, Goldman S, Peberdy MA, Yancy C, Thaneemit-Chen S, Larsen RL, Young J, Lowes B, Rosenberg YD, Investigators BEST (2003) Hormone replacement therapy is associated with improved survival in women with advanced heart failure. J Am Coll Cardiol 42:1238–1245. https://doi.org/10.1016/s0735-1097(03)00938-0
Link S, Meissner M, Held B, Beck A, Weissgerber P, Freichel M, Flockerzi V (2009) Diversity and developmental expression of L-type calcium channel β2 proteins and their influence on calcium current in murine heart *. J Biol Chem 284:30129–30137. https://doi.org/10.1074/jbc.M109.045583
Liu A, Tang M, Xi J, Gao L, Zheng Y, Luo H, Hu X, Zhao F, Reppel M, Hescheler J, Liang H (2010) Functional characterization of inward rectifier potassium ion channel in murine fetal ventricular cardiomyocytes. CPB 26:413–420. https://doi.org/10.1159/000320565
Liu J, Fu JD, Siu CW, Li RA (2007) Functional sarcoplasmic reticulum for calcium handling of human embryonic stem cell-derived cardiomyocytes: insights for driven maturation. Stem Cells 25:3038–3044. https://doi.org/10.1634/stemcells.2007-0549
Liu L, Klein L, Eaton C, Panjrath G, Martin LW, Chae CU, Greenland P, Lloyd-Jones DM, Wactawski-Wende J, Manson JE (2020) Menopausal hormone therapy and risks of first hospitalized heart failure and its subtypes during the intervention and extended postintervention follow-up of the women’s health initiative randomized trials. J Card Fail 26:2–12. https://doi.org/10.1016/j.cardfail.2019.09.006
Liu Y, Liu Y, Li G, Chen Z, Gu G (2018) Ghrelin protects the myocardium with hypoxia/reoxygenation treatment through upregulating the expression of growth hormone, growth hormone secretagogue receptor and insulin-like growth factor-1, and promoting the phosphorylation of protein kinase B. Int J Mol Med 42:3037–3046. https://doi.org/10.3892/ijmm.2018.3886
Lopaschuk GD, Jaswal JS (2010) Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol 56:130–140. https://doi.org/10.1097/FJC.0b013e3181e74a14
Lumbers ER, Boyce AC, Joulianos G, Kumarasamy V, Barner E, Segar JL, Burrell JH (2005) Effects of cortisol on cardiac myocytes and on expression of cardiac genes in fetal sheep. Am J Physiol-Regul Integr Comp Physiol 288:R567–R574. https://doi.org/10.1152/ajpregu.00556.2004
Lundy SD, Zhu W-Z, Regnier M, Laflamme MA (2013) Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev 22:1991–2002. https://doi.org/10.1089/scd.2012.0490
Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ, Kolaja KL, Swanson BJ, January CT (2011) High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol 301:H2006–H2017. https://doi.org/10.1152/ajpheart.00694.2011
Magyar J, Iost N, Körtvély Á, Bányász T, Virág L, Szigligeti P, Varró A, Opincariu M, Szécsi J, Papp JG, Nánási PP (2000) Effects of endothelin-1 on calcium and potassium currents in undiseased human ventricular myocytes. Pflügers Arch Eur J Physiol 441:144–149. https://doi.org/10.1007/s004240000400
Marsh JD, Lehmann MH, Ritchie RH, Gwathmey JK, Green GE, Schiebinger RJ (1998) Androgen receptors mediate hypertrophy in cardiac myocytes. Circulation 98:256–261. https://doi.org/10.1161/01.CIR.98.3.256
Mattingly KA, Ivanova MM, Riggs KA, Wickramasinghe NS, Barch MJ, Klinge CM (2008) Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol Endocrinol 22:609–622. https://doi.org/10.1210/me.2007-0029
McMullen JR, Shioi T, Huang W-Y, Zhang L, Tarnavski O, Bisping E, Schinke M, Kong S, Sherwood MC, Brown J, Riggi L, Kang PM, Izumo S (2004) The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110α) pathway *. J Biol Chem 279:4782–4793. https://doi.org/10.1074/jbc.M310405200
Mendel CM (1989) The free hormone hypothesis: a physiologically based mathematical model*. Endocr Rev 10:232–274. https://doi.org/10.1210/edrv-10-3-232
Menendez-Montes I, Escobar B, Palacios B, Gómez MJ, Izquierdo-Garcia JL, Flores L, Jiménez-Borreguero LJ, Aragones J, Ruiz-Cabello J, Torres M, Martin-Puig S (2016) Myocardial VHL-HIF signaling controls an embryonic metabolic switch essential for cardiac maturation. Dev Cell 39:724–739. https://doi.org/10.1016/j.devcel.2016.11.012
Miki K, Deguchi K, Nakanishi-Koakutsu M, Lucena-Cacace A, Kondo S, Fujiwara Y, Hatani T, Sasaki M, Naka Y, Okubo C, Narita M, Takei I, Napier SC, Sugo T, Imaichi S, Monjo T, Ando T, Tamura N, Imahashi K, Nishimoto T, Yoshida Y (2021) ERRγ enhances cardiac maturation with T-tubule formation in human iPSC-derived cardiomyocytes. Nat Commun 12:3596. https://doi.org/10.1038/s41467-021-23816-3
Mills RJ, Titmarsh DM, Koenig X, Parker BL, Ryall JG, Quaife-Ryan GA, Voges HK, Hodson MP, Ferguson C, Drowley L, Plowright AT, Needham EJ, Wang Q-D, Gregorevic P, Xin M, Thomas WG, Parton RG, Nielsen LK, Launikonis BS, James DE, Elliott DA, Porrello ER, Hudson JE (2017) Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc Natl Acad Sci 114:E8372–E8381. https://doi.org/10.1073/pnas.1707316114
Miracle X, Di Renzo GC, Stark A, Fanaroff A, Carbonell-Estrany X, Saling E, Coordinators Of World Associatin of Perinatal Medicine Prematurity Working Group (2008) Guideline for the use of antenatal corticosteroids for fetal maturation. J Perinat Med 36:191–196. https://doi.org/10.1515/JPM.2008.032
Mitchell JE, Hellkamp AS, Mark DB, Anderson J, Johnson GW, Poole JE, Lee KL, Bardy GH (2013) Thyroid function in heart failure and impact on mortality. JACC Heart Fail 1:48–55. https://doi.org/10.1016/j.jchf.2012.10.004
Mizuno M, Takeba Y, Matsumoto N, Tsuzuki Y, Asoh K, Takagi M, Kobayashi S, Yamamoto H (2010) Antenatal glucocorticoid therapy accelerates ATP production with creatine kinase increase in the growth-enhanced fetal rat heart. Circ J 74:171–180. https://doi.org/10.1253/circj.CJ-09-0311
Montessuit C, Palma T, Viglino C, Pellieux C, Lerch R (2006) Effects of insulin-like growth factor-I on the maturation of metabolism in neonatal rat cardiomyocytes. Pflugers Arch Eur J Physiol 452:380–386. https://doi.org/10.1007/s00424-006-0059-4
Moreno-Domínguez A, Ortega-Sáenz P, Gao L, Colinas O, García-Flores P, Bonilla-Henao V, Aragonés J, Hüttemann M, Grossman LI, Weissmann N, Sommer N, López-Barneo J (2020) Acute O2 sensing through HIF2α-dependent expression of atypical cytochrome oxidase subunits in arterial chemoreceptors. Sci Signal 13:eaay9452. https://doi.org/10.1126/scisignal.aay9452
Mukoyama M, Nakao K, Hosoda K, Suga S, Saito Y, Ogawa Y, Shirakami G, Jougasaki M, Obata K, Yasue H (1991) Brain natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J Clin Invest 87:1402–1412. https://doi.org/10.1172/JCI115146
Mulieri LA, Hasenfuss G, Leavitt B, Allen PD, Alpert NR (1992) Altered myocardial force-frequency relation in human heart failure. Circulation 85:1743–1750. https://doi.org/10.1161/01.cir.85.5.1743
Murphy SA, Chen EZ, Tung L, Boheler KR, Kwon C (2021) Maturing heart muscle cells: mechanisms and transcriptomic insights. Semin Cell Dev Biol 119:49–60. https://doi.org/10.1016/j.semcdb.2021.04.019
Murphy SA, Miyamoto M, Kervadec A, Kannan S, Tampakakis E, Kambhampati S, Lin BL, Paek S, Andersen P, Lee D-I, Zhu R, An SS, Kass DA, Uosaki H, Colas AR, Kwon C (2021) PGC1/PPAR drive cardiomyocyte maturation at single cell level via YAP1 and SF3B2. Nat Commun 12:1648. https://doi.org/10.1038/s41467-021-21957-z
Nakano H, Minami I, Braas D, Pappoe H, Wu X, Sagadevan A, Vergnes L, Fu K, Morselli M, Dunham C, Ding X, Stieg AZ, Gimzewski JK, Pellegrini M, Clark PM, Reue K, Lusis AJ, Ribalet B, Kurdistani SK, Christofk H, Nakatsuji N, Nakano A (2017) Glucose inhibits cardiac muscle maturation through nucleotide biosynthesis. Elife 6:e29330. https://doi.org/10.7554/eLife.29330
Naqvi N, Li M, Calvert JW, Tejada T, Lambert JP, Wu J, Kesteven SH, Holman SR, Matsuda T, Lovelock JD, Howard WW, Iismaa SE, Chan AY, Crawford BH, Wagner MB, Martin DIK, Lefer DJ, Graham RM, Husain A (2014) A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell 157:795–807. https://doi.org/10.1016/j.cell.2014.03.035
Nerbonne JM, Kass RS (2005) Molecular physiology of cardiac repolarization. Physiol Rev 85:1205–1253. https://doi.org/10.1152/physrev.00002.2005
Nishida S, Matsumura S, Horino M, Oyama H, Tenku A (1977) The variations of plasma corticosterone/cortisol ratios following ACTH stimulation or dexamethasone administration in normal men. J Clin Endocrinol Metab 45:585–588. https://doi.org/10.1210/jcem-45-3-585
Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, Jiang J, Massé S, Gagliardi M, Hsieh A, Thavandiran N, Laflamme MA, Nanthakumar K, Gross GJ, Backx PH, Keller G, Radisic M (2013) Biowire: a platform for maturation of human pluripotent stem cell–derived cardiomyocytes. Nat Methods 10:781–787. https://doi.org/10.1038/nmeth.2524
Ogawa T, de Bold AJ (2014) The heart as an endocrine organ. Endocr Connect 3:R31–R44. https://doi.org/10.1530/EC-14-0012
Oka Y, Rozek LM, Czech MP (1985) Direct demonstration of rapid insulin-like growth factor II Receptor internalization and recycling in rat adipocytes. Insulin stimulates 125I-insulin-like growth factor II degradation by modulating the IGF-II receptor recycling process. J Biol Chem 260:9435–9442
Olivetti G, Cigola E, Maestri R, Corradi D, Lagrasta C, Gambert SR, Anversa P (1996) Aging, cardiac hypertrophy and ischemic cardiomyopathy do not affect the proportion of mononucleated and multinucleated myocytes in the human heart. J Mol Cell Cardiol 28:1463–1477. https://doi.org/10.1006/jmcc.1996.0137
Ono K, Iijima T (2010) Cardiac T-type Ca(2+) channels in the heart. J Mol Cell Cardiol 48:65–70. https://doi.org/10.1016/j.yjmcc.2009.08.021
Page E, McCallister LP (1973) Quantitative electron microscopic description of heart muscle cells: Application to normal, hypertrophied and thyroxin-stimulated hearts. Am J Cardiol 31:172–181. https://doi.org/10.1016/0002-9149(73)91030-8
Paigel AS, Ribeiro RF, Fernandes AA, Targueta GP, Vassallo DV, Stefanon I (2011) Myocardial contractility is preserved early but reduced late after ovariectomy in young female rats. Reprod Biol Endocrinol 9:54. https://doi.org/10.1186/1477-7827-9-54
Pandit S, Woranush W, Wattanapermpool J, Bupha-Intr T (2014) Significant role of female sex hormones in cardiac myofilament activation in angiotensin II-mediated hypertensive rats. J Physiol Sci 64:269–277. https://doi.org/10.1007/s12576-014-0316-9
Pantos C, Malliopoulou V, Mourouzis I, Karamanoli E, Moraitis P, Tzeis S, Paizis I, Cokkinos AD, Carageorgiou H, Varonos DD, Cokkinos DV (2003) Thyroxine pretreatment increases basal myocardial heat-shock protein 27 expression and accelerates translocation and phosphorylation of this protein upon ischaemia. Eur J Pharmacol 478:53–60. https://doi.org/10.1016/j.ejphar.2003.08.030
Pantos C, Malliopoulou V, Paizis I, Moraitis P, Mourouzis I, Tzeis S, Karamanoli E, Cokkinos DD, Carageorgiou H, Varonos D, Cokkinos DV (2003) Thyroid hormone and cardioprotection: study of p38 MAPK and JNKs during ischaemia and at reperfusion in isolated rat heart. Mol Cell Biochem 242:173–180
Pantos C, Mourouzis I, Saranteas T, Brozou V, Galanopoulos G, Kostopanagiotou G, Cokkinos DV (2011) Acute T3 treatment protects the heart against ischemia-reperfusion injury via TRα1 receptor. Mol Cell Biochem 353:235. https://doi.org/10.1007/s11010-011-0791-8
Pantos C, Mourouzis I, Saranteas T, Clavé G, Ligeret H, Noack-Fraissignes P, Renard P-Y, Massonneau M, Perimenis P, Spanou D, Kostopanagiotou G, Cokkinos DV (2009) Thyroid hormone improves postischaemic recovery of function while limiting apoptosis: a new therapeutic approach to support hemodynamics in the setting of ischaemia-reperfusion? Basic Res Cardiol 104:69–77. https://doi.org/10.1007/s00395-008-0758-4
Pantos CI, Malliopoulou VA, Mourouzis IS, Karamanoli EP, Tzeis SM, Carageorgiou HC, Varonos DD, Cokkinos DV (2001) Long-term thyroxine administration increases heat stress protein-70 mRNA expression and attenuates p38 MAP kinase activity in response to ischaemia. J Endocrinol 170:207–215. https://doi.org/10.1677/joe.0.1700207
Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, Walsh K (2012) Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res 111:1012–1026. https://doi.org/10.1161/CIRCRESAHA.112.274142
Papp R, Bett GCL, Lis A, Rasmusson RL, Baczkó I, Varró A, Salama G (2017) Genomic upregulation of cardiac Cav1.2α and NCX1 by estrogen in women. Biol Sex Differ 8:26. https://doi.org/10.1186/s13293-017-0148-4
Paredes A, Justo-Méndez R, Jiménez-Blasco D, Núñez V, Calero I, Villalba-Orero M, Alegre-Martí A, Fischer T, Gradillas A, Sant’Anna VAR, Were F, Huang Z, Hernansanz-Agustín P, Contreras C, Martínez F, Camafeita E, Vázquez J, Ruiz-Cabello J, Area-Gómez E, Sánchez-Cabo F, Treuter E, Bolaños JP, Estébanez-Perpiñá E, Rupérez FJ, Barbas C, Enríquez JA, Ricote M (2023) γ-Linolenic acid in maternal milk drives cardiac metabolic maturation. Nature. https://doi.org/10.1038/s41586-023-06068-7
Parikh SS, Blackwell DJ, Gomez-Hurtado N, Frisk M, Wang L, Kim K, Dahl CP, Fiane A, Tønnessen T, Kryshtal DO, Louch WE, Knollmann BC (2017) Thyroid and glucocorticoid hormones promote functional T-tubule development in human-induced pluripotent stem cell derived cardiomyocytes. Circ Res 121:1323. https://doi.org/10.1161/CIRCRESAHA.117.311920
Patel S, Rauf A, Khan H, Abu-Izneid T (2017) Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed Pharmacother 94:317–325. https://doi.org/10.1016/j.biopha.2017.07.091
Patel SP, Campbell DL (2005) Transient outward potassium current, ‘Ito’, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J Physiol 569:7–39. https://doi.org/10.1113/jphysiol.2005.086223
Patten RD, Karas RH (2006) Estrogen replacement and cardiomyocyte protection. Trends Cardiovasc Med 16:69–75. https://doi.org/10.1016/j.tcm.2006.01.002
Patterson E, Ma L, Szabo B, Robinson CP, Thadani U (1998) Ovariectomy and estrogen-induced alterations in myocardial contractility in female rabbits: role of the L-type calcium channel. J Pharmacol Exp Ther 284:586–591
Patterson M, Barske L, Van Handel B, Rau CD, Gan P, Sharma A, Parikh S, Denholtz M, Huang Y, Yamaguchi Y, Shen H, Allayee H, Crump JG, Force TI, Lien C-L, Makita T, Lusis AJ, Kumar SR, Sucov HM (2017) Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat Genet 49:1346–1353. https://doi.org/10.1038/ng.3929
Pedernera E, Gómora MJ, Meneses I, Ita MD, Méndez C (2017) Androgen receptor is expressed in mouse cardiomyocytes at prenatal and early postnatal developmental stages. BMC Physiol. https://doi.org/10.1186/s12899-017-0033-8
Peinkofer G, Burkert K, Urban K, Krausgrill B, Hescheler J, Saric T, Halbach M (2016) From early embryonic to adult stage: comparative study of action potentials of native and pluripotent stem cell-derived cardiomyocytes. Stem Cells Dev 25:1397–1406. https://doi.org/10.1089/scd.2016.0073
Pongkan W, Chattipakorn SC, Chattipakorn N (2016) Roles of testosterone replacement in cardiac ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther 21:27–43. https://doi.org/10.1177/1074248415587977
Poon E, Keung W, Liang Y, Ramalingam R, Yan B, Zhang S, Chopra A, Moore J, Herren A, Lieu DK, Wong HS, Weng Z, Wong OT, Lam YW, Tomaselli GF, Chen C, Boheler KR, Li RA (2015) Proteomic analysis of human pluripotent stem cell-derived, fetal, and adult ventricular cardiomyocytes reveals pathways crucial for cardiac metabolism and maturation. Circ Cardiovasc Genet 8:427–436. https://doi.org/10.1161/CIRCGENETICS.114.000918
Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA (2011) Transient regenerative potential of the neonatal mouse heart. Science 331:1078–1080. https://doi.org/10.1126/science.1200708
Portman MA, Slee A, Olson AK, Cohen G, Karl T, Tong E, Hastings L, Patel H, Reinhartz O, Mott AR, Mainwaring R, Linam J, Danzi S, Investigators TRICC (2010) Triiodothyronine Supplementation in Infants and Children Undergoing Cardiopulmonary Bypass (TRICC): a multicenter placebo-controlled randomized trial: age analysis. Circulation 122:S224-233. https://doi.org/10.1161/CIRCULATIONAHA.109.926394
Psarra A-MG, Solakidi S, Sekeris CE (2006) The mitochondrion as a primary site of action of steroid and thyroid hormones: Presence and action of steroid and thyroid hormone receptors in mitochondria of animal cells. Mol Cell Endocrinol 246:21–33. https://doi.org/10.1016/j.mce.2005.11.025
Qu Y, Boutjdir M (2001) Gene expression of SERCA2a and L- and T-type Ca channels during human heart development. Pediatr Res 50:569–574. https://doi.org/10.1203/00006450-200111000-00006
Quinn SJ, Williams GH (1988) Regulation of aldosterone secretion. Annu Rev Physiol 50:409–426. https://doi.org/10.1146/annurev.ph.50.030188.002205
Radisic M, Park H, Shing H, Consi T, Schoen FJ, Langer R, Freed LE, Vunjak-Novakovic G (2004) Functional assembly of engineered myocardium by electrical stimulation of cardiac myocytes cultured on scaffolds. Proc Natl Acad Sci 101:18129–18134. https://doi.org/10.1073/pnas.0407817101
Ranasinghe AM, Quinn DW, Pagano D, Edwards N, Faroqui M, Graham TR, Keogh BE, Mascaro J, Riddington DW, Rooney SJ, Townend JN, Wilson IC, Bonser RS (2006) Glucose-insulin-potassium and triiodothyronine individually improve hemodynamic performance and are associated with reduced troponin I release after on-pump coronary artery bypass grafting. Circulation 114:I245-250. https://doi.org/10.1161/CIRCULATIONAHA.105.000786
Ren J, Brown-Borg HM (2002) Impaired cardiac excitation-contraction coupling in ventricular myocytes from Ames dwarf mice with IGF-I deficiency. Growth Horm IGF Res 12:99–105. https://doi.org/10.1054/ghir.2002.0267
Rhee SS, Pearce EN (2011) The endocrine system and the heart: a review. Rev Esp Cardiol 64:220–231. https://doi.org/10.1016/j.rec.2010.10.016
Ribeiro MC, Tertoolen LG, Guadix JA, Bellin M, Kosmidis G, D’Aniello C, Monshouwer-Kloots J, Goumans M-J, Wang Y, Feinberg AW, Mummery CL, Passier R (2015) Functional maturation of human pluripotent stem cell derived cardiomyocytes in vitro—correlation between contraction force and electrophysiology. Biomaterials 51:138–150. https://doi.org/10.1016/j.biomaterials.2015.01.067
Rog-Zielinska EA, Craig M-A, Manning JR, Richardson RV, Gowans GJ, Dunbar DR, Gharbi K, Kenyon CJ, Holmes MC, Hardie DG, Smith GL, Chapman KE (2015) Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: a role for PGC-1α. Cell Death Differ 22:1106–1116. https://doi.org/10.1038/cdd.2014.181
Rog-Zielinska EA, Thomson A, Kenyon CJ, Brownstein DG, Moran CM, Szumska D, Michailidou Z, Richardson J, Owen E, Watt A, Morrison H, Forrester LM, Bhattacharya S, Holmes MC, Chapman KE (2013) Glucocorticoid receptor is required for foetal heart maturation. Hum Mol Genet 22:3269–3282. https://doi.org/10.1093/hmg/ddt182
Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, Morikawa K, Teles D, Yazawa M, Vunjak-Novakovic G (2018) Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556:239–243. https://doi.org/10.1038/s41586-018-0016-3
Ropero AB, Eghbali M, Minosyan TY, Tang G, Toro L, Stefani E (2006) Heart estrogen receptor alpha: distinct membrane and nuclear distribution patterns and regulation by estrogen. J Mol Cell Cardiol 41:496–510. https://doi.org/10.1016/j.yjmcc.2006.05.022
Rozanski A, Takano APC, Kato PN, Soares AG, Lellis-Santos C, Campos JC, Ferreira JCB, Barreto-Chaves MLM, Moriscot AS (2013) M-protein is down-regulated in cardiac hypertrophy driven by thyroid hormone in rats. Mol Endocrinol 27:2055–2065. https://doi.org/10.1210/me.2013-1018
Sakaguchi Y, Cui G, Sen L (1996) Acute effects of thyroid hormone on inward rectifier potassium channel currents in guinea pig ventricular myocytes. Endocrinology 137:4744–4751. https://doi.org/10.1210/endo.137.11.8895342
Sakamoto T, Batmanov K, Wan S, Guo Y, Lai L, Vega RB, Kelly DP (2022) The nuclear receptor ERR cooperates with the cardiogenic factor GATA4 to orchestrate cardiomyocyte maturation. Nat Commun 13:1991. https://doi.org/10.1038/s41467-022-29733-3
Sakamoto T, Matsuura TR, Wan S, Ryba DM, Kim J, Won KJ, Lai L, Petucci C, Petrenko N, Musunuru K, Vega RB, Kelly DP (2020) A critical role for estrogen-related receptor signaling in cardiac maturation. Circ Res 126:1685–1702. https://doi.org/10.1161/CIRCRESAHA.119.316100
dos Santos RL, da Silva FB, Ribeiro RF, Stefanon I (2014) Sex hormones in the cardiovascular system. Horm Mol Biol Clin Invest 18:89–103. https://doi.org/10.1515/hmbci-2013-0048
Sbert-Roig M, Bauzá-Thorbrügge M, Galmés-Pascual BM, Capllonch-Amer G, García-Palmer FJ, Lladó I, Proenza AM, Gianotti M (2016) GPER mediates the effects of 17β-estradiol in cardiac mitochondrial biogenesis and function. Mol Cell Endocrinol 420:116–124. https://doi.org/10.1016/j.mce.2015.11.027
Schaible TF, Malhotra A, Ciambrone G, Scheuer J (1984) The effects of gonadectomy on left ventricular function and cardiac contractile proteins in male and female rats. Circ Res 54:38–49. https://doi.org/10.1161/01.res.54.1.38
Schaper J, Meiser E, Stämmler G (1985) Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ Res 56:377–391. https://doi.org/10.1161/01.RES.56.3.377
Schelling JR, DeLuca DJ, Konieczkowski M, Marzec R, Sedor JR, Dubyak GR, Linas SL (1994) Glucocorticoid uncoupling of antiogensin II-dependent phospholipase C activation in rat vascular smooth muscle cells. Kidney Int 46:675–682. https://doi.org/10.1038/ki.1994.320
Schiaffino S, Gorza L, Ausoni S (1993) Troponin isoform switching in the developing heart and its functional consequences. Trends Cardiovasc Med 3:12–17. https://doi.org/10.1016/1050-1738(93)90022-X
Schierbeck LL, Rejnmark L, Tofteng CL, Stilgren L, Eiken P, Mosekilde L, Køber L, Jensen J-EB (2012) Effect of hormone replacement therapy on cardiovascular events in recently post-menopausal women: randomised trial. BMJ 345:e6409. https://doi.org/10.1136/bmj.e6409
Schuster I, Mahmoodzadeh S, Dworatzek E, Jaisser F, Messaoudi S, Morano I, Regitz-Zagrosek V (2016) Cardiomyocyte-specific overexpression of oestrogen receptor β improves survival and cardiac function after myocardial infarction in female and male mice. Clin Sci (Lond) 130:365–376. https://doi.org/10.1042/CS20150609
Semenza GL, Jiang B-H, Leung SW, Passantino R, Concordet J-P, Maire P, Giallongo A (1996) hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1*. J Biol Chem 271:32529–32537. https://doi.org/10.1074/jbc.271.51.32529
Sensky PL, Roy CH, Barnes RJ, Heath MF (1994) Changes in fetal thyroid hormone levels in adrenalectomized fetal sheep following continuous cortisol infusion 72 h before delivery. J Endocrinol 140:79–83. https://doi.org/10.1677/joe.0.1400079
Seppet EK, Kaambre T, Sikk P, Tiivel T, Vija H, Tonkonogi M, Sahlin K, Kay L, Appaix F, Braun U, Eimre M, Saks VA (2001) Functional complexes of mitochondria with Ca, MgATPases of myofibrils and sarcoplasmic reticulum in muscle cells. Biochim Biophys Acta (BBA)-Bioenergetics 1504:379–395. https://doi.org/10.1016/S0005-2728(00)00269-3
Setterberg IE, Le C, Frisk M, Perdreau-Dahl H, Li J, Louch WE (2021) The physiology and pathophysiology of t-tubules in the heart. Front Physiol. https://doi.org/10.3389/fphys.2021.718404
Shen H, Gan P, Wang K, Darehzereshki A, Wang K, Kumar SR, Lien C-L, Patterson M, Tao G, Sucov HM (2020) Mononuclear diploid cardiomyocytes support neonatal mouse heart regeneration in response to paracrine IGF2 signaling. Elife 9:e53071. https://doi.org/10.7554/eLife.53071
Shepard TH, Muffley LA, Thayer Smith L (1998) Ultrastructural study of mitochondria and their cristae in embryonic rats and primate (N. nemistrina). Anat Rec 252:383–392. https://doi.org/10.1002/(SICI)1097-0185(199811)252:3%3c383::AID-AR6%3e3.0.CO;2-Z
Shlipak MG, Angeja BG, Go AS, Frederick PD, Canto JG, Grady D (2001) Hormone therapy and in-hospital survival after myocardial infarction in postmenopausal women. Circulation 104:2300–2304. https://doi.org/10.1161/hc4401.98414
Silver M, Fowden AL (1988) Induction of labour in domestic animals: endocrine changes and neonatal viability. In: Künzel W, Jensen A (eds) The endocrine control of the fetus. Springer, Berlin, pp 401–411
Sim CB, Phipson B, Ziemann M, Rafehi H, Mills RJ, Watt KI, Abu-Bonsrah KD, Kalathur RKR, Voges HK, Dinh DT, ter Huurne M, Vivien CJ, Kaspi A, Kaipananickal H, Hidalgo A, Delbridge LMD, Robker RL, Gregorevic P, dos Remedios CG, Lal S, Piers AT, Konstantinov IE, Elliott DA, El-Osta A, Oshlack A, Hudson JE, Porrello ER (2021) Sex-specific control of human heart maturation by the progesterone receptor. Circulation 143:1614–1628. https://doi.org/10.1161/CIRCULATIONAHA.120.051921
Song R, Hu X-Q, Zhang L (2019) Glucocorticoids and programming of the microenvironment in heart. J Endocrinol 242:T121–T133. https://doi.org/10.1530/JOE-18-0672
Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ (1996) Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol-Heart Circ Physiol 271:H2183–H2189. https://doi.org/10.1152/ajpheart.1996.271.5.H2183
Soonpaa MH, Zebrowski DC, Platt C, Rosenzweig A, Engel FB, Field LJ (2015) Cardiomyocyte cell-cycle activity during preadolescence. Cell 163:781–782. https://doi.org/10.1016/j.cell.2015.10.037
Spach MS, Heidlage JF, Barr RC, Dolber PC (2004) Cell size and communication: role in structural and electrical development and remodeling of the heart. Heart Rhythm 1:500–515. https://doi.org/10.1016/j.hrthm.2004.06.010
Spach MS, Heidlage JF, Dolber PC, Barr RC (2000) Electrophysiological effects of remodeling cardiac gap junctions and cell size: experimental and model studies of normal cardiac growth. Circ Res 86:302–311. https://doi.org/10.1161/01.res.86.3.302
Spinedi E, Aguado L, Basilotta G, Carrizo D (1989) Angiotensin II and glucocorticoid release: direct effect at the adrenal level and modulation of the adrenocorticotropin-induced glucocorticoid release. J Endocrinol Invest 12:321–327. https://doi.org/10.1007/BF03349997
Steinmetz M, Quentin Th, Poppe A, Paul Th, Jux Ch (2005) Changes in expression levels of genes involved in fatty acid metabolism: upregulation of all three members of the PPAR family (α, γ, δ) and the newly described adiponectin receptor 2, but not adiponectin receptor 1 during neonatal cardiac development of the rat. Basic Res Cardiol 100:263–269. https://doi.org/10.1007/s00395-005-0520-0
Stock A, Sies H (2000) Thyroid hormone receptors bind to an element in the connexin43 promoter. Biol Chem 381:973–979. https://doi.org/10.1515/BC.2000.120
Sudoh T, Kangawa K, Minamino N, Matsuo H (1988) A new natriuretic peptide in porcine brain. Nature 332:78–81. https://doi.org/10.1038/332078a0
Suleiman M-S, Singh RJR, Stewart CEH (2007) Apoptosis and the cardiac action of insulin-like growth factor I. Pharmacol Ther 114:278–294. https://doi.org/10.1016/j.pharmthera.2007.03.001
Sun ZQ, Ojamaa K, Nakamura TY, Artman M, Klein I, Coetzee WA (2001) Thyroid hormone increases pacemaker activity in rat neonatal atrial myocytes. J Mol Cell Cardiol 33:811–824. https://doi.org/10.1006/jmcc.2001.1353
Sundgren NC, Giraud GD, Schultz JM, Lasarev MR, Stork PJS, Thornburg KL (2003) Extracellular signal-regulated kinase and phosphoinositol-3 kinase mediate IGF-1 induced proliferation of fetal sheep cardiomyocytes. Am J Physiol Regul Integr Comp Physiol 285:R1481-1489. https://doi.org/10.1152/ajpregu.00232.2003
Suzuki T, Kumazaki T, Mitsui Y (1993) Endothelin-1 is produced and secreted by neonatal rat cardiac myocytes in vitro. Biochem Biophys Res Commun 191:823–830. https://doi.org/10.1006/bbrc.1993.1291
Svensson Holm AB, Lindgren I, Österman H, Altimiras J (2014) Thyroid hormone does not induce maturation of embryonic chicken cardiomyocytes in vitro. Physiol Rep 2:e12182. https://doi.org/10.14814/phy2.12182
Synnergren J, Améen C, Jansson A, Sartipy P (2012) Global transcriptional profiling reveals similarities and differences between human stem cell-derived cardiomyocyte clusters and heart tissue. Physiol Genomics 44:245–258. https://doi.org/10.1152/physiolgenomics.00118.2011
Tajima M, Weinberg EO, Bartunek J, Jin H, Yang R, Paoni NF, Lorell BH (1999) Treatment with growth hormone enhances contractile reserve and intracellular calcium transients in myocytes from rats with postinfarction heart failure. Circulation 99:127–134. https://doi.org/10.1161/01.CIR.99.1.127
Takimoto E (2012) Cyclic GMP-dependent signaling in cardiac myocytes. Circ J 76:1819–1825. https://doi.org/10.1253/circj.CJ-12-0664
Tan L, Bogush N, Naib H, Perry J, Calvert JW, Martin DIK, Graham RM, Naqvi N, Husain A (2019) Redox activation of JNK2α2 mediates thyroid hormone-stimulated proliferation of neonatal murine cardiomyocytes. Sci Rep 9:17731. https://doi.org/10.1038/s41598-019-53705-1
Tang H-Y, Lin H-Y, Zhang S, Davis FB, Davis PJ (2004) Thyroid hormone causes mitogen-activated protein kinase-dependent phosphorylation of the nuclear estrogen receptor. Endocrinology 145:3265–3272. https://doi.org/10.1210/en.2004-0308
Teng Z, Zhang M, Zhao M, Zhang W (2013) Glucocorticoid exerts its non-genomic effect on IPSC by activation of a phospholipase C-dependent pathway in prefrontal cortex of rats. J Physiol 591:3341–3353. https://doi.org/10.1113/jphysiol.2013.254961
Thomas LW, Ashcroft M (2019) Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria. Cell Mol Life Sci 76:1759–1777. https://doi.org/10.1007/s00018-019-03039-y
Touitou Y, Auzeby A, Bogdan A (1990) Cortisol and cortisone production in rat and mouse adrenal incubations. J Steroid Biochem Mol Biol 37:279–284. https://doi.org/10.1016/0960-0760(90)90339-m
Tower J (2006) Sex-specific regulation of aging and apoptosis. Mech Ageing Dev 127:705–718. https://doi.org/10.1016/j.mad.2006.05.001
Troncoso R, Ibarra C, Vicencio JM, Jaimovich E, Lavandero S (2014) New insights into IGF-1 signaling in the heart. Trends Endocrinol Metab 25:128–137. https://doi.org/10.1016/j.tem.2013.12.002
Turdi S, Huff AF, Pang J, He EY, Chen X, Wang S, Chen Y, Zhang Y, Ren J (2015) 17-β Estradiol attenuates ovariectomy-induced changes in cardiomyocyte contractile function via activation of AMP-activated protein kinase. Toxicol Lett 232:253–262. https://doi.org/10.1016/j.toxlet.2014.11.012
van Tuyl M, Blommaart PE, de Boer PAJ, Wert SE, Ruijter JM, Islam S, Schnitzer J, Ellison AR, Tibboel D, Moorman AFM, Lamers WH (2004) Prenatal exposure to thyroid hormone is necessary for normal postnatal development of murine heart and lungs. Dev Biol 272:104–117. https://doi.org/10.1016/j.ydbio.2004.03.042
Underwood RH, Williams GH (1972) The simultaneous measurement of aldosterone, cortisol, and corticosterone in human peripheral plasma by displacement analysis. J Lab Clin Med 79:848–862. https://doi.org/10.5555/uri:pii:0022214372900558
Uosaki H, Cahan P, Lee DI, Wang S, Miyamoto M, Fernandez L, Kass DA, Kwon C (2015) Transcriptional landscape of cardiomyocyte maturation. Cell Rep 13:1705–1716. https://doi.org/10.1016/j.celrep.2015.10.032
Vandenplas G, De Bacquer D, Calders P, Fiers T, Kaufman J-M, Ouwens DM, Ruige JB (2012) Endogenous oestradiol and cardiovascular disease in healthy men: a systematic review and meta-analysis of prospective studies. Heart 98:1478–1482. https://doi.org/10.1136/heartjnl-2011-301587
VanDusen NJ, Lee JY, Gu W, Butler CE, Sethi I, Zheng Y, King JS, Zhou P, Suo S, Guo Y, Ma Q, Yuan G-C, Pu WT (2021) Massively parallel in vivo CRISPR screening identifies RNF20/40 as epigenetic regulators of cardiomyocyte maturation. Nat Commun 12:4442. https://doi.org/10.1038/s41467-021-24743-z
Vasan RS, Sullivan LM, D’Agostino RB, Roubenoff R, Harris T, Sawyer DB, Levy D, Wilson PWF (2003) Serum insulin-like growth factor I and risk for heart failure in elderly individuals without a previous myocardial infarction: the Framingham Heart Study. Ann Intern Med 139:642–648. https://doi.org/10.7326/0003-4819-139-8-200310210-00007
Veerman CC, Kosmidis G, Mummery CL, Casini S, Verkerk AO, Bellin M (2015) Immaturity of human stem-cell-derived cardiomyocytes in culture: fatal flaw or soluble problem? Stem Cells Dev 24:1035–1052. https://doi.org/10.1089/scd.2014.0533
Ventura-Clapier R, Moulin M, Piquereau J, Lemaire C, Mericskay M, Veksler V, Garnier A (2017) Mitochondria: a central target for sex differences in pathologies. Clin Sci (Lond) 131:803–822. https://doi.org/10.1042/CS20160485
Verkerk AO, Wilders R, Veldkamp MW, de Geringel W, Kirkels JH, Tan HL (2005) Gender disparities in cardiac cellular electrophysiology and arrhythmia susceptibility in human failing ventricular myocytes. Int Heart J 46:1105–1118. https://doi.org/10.1536/ihj.46.1105
Vijay V, Han T, Moland CL, Kwekel JC, Fuscoe JC, Desai VG (2015) Sexual dimorphism in the expression of mitochondria-related genes in rat heart at different ages. PLoS ONE 10:e0117047. https://doi.org/10.1371/journal.pone.0117047
Vila G, Grimm G, Resl M, Heinisch B, Einwallner E, Esterbauer H, Dieplinger B, Mueller T, Luger A, Clodi M (2012) B-type natriuretic peptide modulates ghrelin, hunger, and satiety in healthy men. Diabetes 61:2592–2596. https://doi.org/10.2337/db11-1466
de Vries WB, van der Leij FR, Bakker JM, Kamphuis PJGH, van Oosterhout MFM, Schipper MEI, Smid GB, Bartelds B, van Bel F (2002) Alterations in adult rat heart after neonatal dexamethasone therapy. Pediatr Res 52:900–906. https://doi.org/10.1203/00006450-200212000-00015
Wadman M (2023) FDA no longer has to require animal testing for new drugs. Science 379:127–128. https://doi.org/10.1126/science.adg6276
Wadthaisong M, Witayavanitkul N, Bupha-Intr T, Wattanapermpool J, de Tombe PP (2019) Chronic high-dose testosterone treatment: impact on rat cardiac contractile biology. Physiolog Rep 7:e14192. https://doi.org/10.14814/phy2.14192
Walklate J, Ferrantini C, Johnson CA, Tesi C, Poggesi C, Geeves MA (2021) Alpha and beta myosin isoforms and human atrial and ventricular contraction. Cell Mol Life Sci 78:7309–7337. https://doi.org/10.1007/s00018-021-03971-y
Wang L, Wada Y, Ballan N, Schmeckpeper J, Huang J, Rau CD, Wang Y, Gepstein L, Knollmann BC (2021) Triiodothyronine and dexamethasone alter potassium channel expression and promote electrophysiological maturation of human-induced pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol 161:130–138. https://doi.org/10.1016/j.yjmcc.2021.08.005
Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, Kelly M, Meldrum DR (2009) Estrogen receptor β mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol 296:R972–R978. https://doi.org/10.1152/ajpregu.00045.2009
Wang T, Liu J, McDonald C, Lupino K, Zhai X, Wilkins BJ, Hakonarson H, Pei L (2017) GDF15 is a heart-derived hormone that regulates body growth. EMBO Mol Med 9:1150–1164. https://doi.org/10.15252/emmm.201707604
Wang WE, Li L, Xia X, Fu W, Liao Q, Lan C, Yang D, Chen H, Yue R, Zeng C, Zhou L, Zhou B, Duan DD, Chen X, Houser SR, Zeng C (2017) Dedifferentiation, proliferation, and redifferentiation of adult mammalian cardiomyocytes after ischemic injury. Circulation 136:834–848. https://doi.org/10.1161/CIRCULATIONAHA.116.024307
Wang Y, Xu H, Kumar R, Tipparaju SM, Wagner MB, Joyner RW (2003) Differences in transient outward current properties between neonatal and adult human atrial myocytes. J Mol Cell Cardiol 35:1083–1092. https://doi.org/10.1016/s0022-2828(03)00200-1
Wattanapermpool J, Reiser PJ (1999) Differential effects of ovariectomy on calcium activation of cardiac and soleus myofilaments. Am J Physiol 277:H467-473. https://doi.org/10.1152/ajpheart.1999.277.2.H467
Wei Y, Li J, Huang J, Zhang X, Zhao H, Cui C, Li Y, Hu S (2012) Elevation of IGF-2 receptor and the possible underlying implications in end-stage heart failure patients before and after heart transplantation. J Cell Mol Med 16:1038–1046. https://doi.org/10.1111/j.1582-4934.2011.01414.x
White P, Burton KA, Fowden AL, Dauncey MJ (2001) Developmental expression analysis of thyroid hormone receptor isoforms reveals new insights into their essential functions in cardiac and skeletal muscles. FASEB J 15:1367–1376. https://doi.org/10.1096/fj.00-0725com
Wickramasinghe NM, Sachs D, Shewale B, Gonzalez DM, Dhanan-Krishnan P, Torre D, LaMarca E, Raimo S, Dariolli R, Serasinghe MN, Mayourian J, Sebra R, Beaumont K, Iyengar S, French DL, Hansen A, Eschenhagen T, Chipuk JE, Sobie EA, Jacobs A, Akbarian S, Ischiropoulos H, Ma’ayanHoutenCostaDubois ASMKNC (2022) PPARdelta activation induces metabolic and contractile maturation of human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 29:559-576.e7. https://doi.org/10.1016/j.stem.2022.02.011
Wiegerinck RF, Cojoc A, Zeidenweber CM, Ding G, Shen M, Joyner RW, Fernandez JD, Kanter KR, Kirshbom PM, Kogon BE, Wagner MB (2009) Force frequency relationship of the human ventricle increases during early postnatal development. Pediatr Res 65:414–419. https://doi.org/10.1203/PDR.0b013e318199093c
Wilson EM, French FS (1976) Binding properties of androgen receptors. Evidence for identical receptors in rat testis, epididymis, and prostate. J Biol Chem 251:5620–5629. https://doi.org/10.1016/S0021-9258(17)33103-4
Wu Y, Peng J, Campbell KB, Labeit S, Granzier H (2007) Hypothyroidism leads to increased collagen-based stiffness and re-expression of large cardiac titin isoforms with high compliance. J Mol Cell Cardiol 42:186–195. https://doi.org/10.1016/j.yjmcc.2006.09.017
Wulf A, Harneit A, Kröger M, Kebenko M, Wetzel MG, Weitzel JM (2008) T3-mediated expression of PGC-1alpha via a far upstream located thyroid hormone response element. Mol Cell Endocrinol 287:90–95. https://doi.org/10.1016/j.mce.2008.01.017
Xu S, Xie F, Tian L, Fallah S, Babaei F, Manno SHC, Manno FAM, Zhu L, Wong KF, Liang Y, Ramalingam R, Sun L, Wang X, Plumb R, Gethings L, Lam YW, Cheng SH (2020) Estrogen accelerates heart regeneration by promoting the inflammatory response in zebrafish. J Endocrinol 245:39–51. https://doi.org/10.1530/JOE-19-0413
Yang X, Rodriguez M, Pabon L, Fischer KA, Reinecke H, Regnier M, Sniadecki NJ, Ruohola-Baker H, Murry CE (2014) Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J Mol Cell Cardiol 72:296–304. https://doi.org/10.1016/j.yjmcc.2014.04.005
Yang X, Rodriguez ML, Leonard A, Sun L, Fischer KA, Wang Y, Ritterhoff J, Zhao L, Kolwicz SC, Pabon L, Reinecke H, Sniadecki NJ, Tian R, Ruohola-Baker H, Xu H, Murry CE (2019) Fatty acids enhance the maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cell Reports 13:657–668. https://doi.org/10.1016/j.stemcr.2019.08.013
Zeng B, Liao X, Liu L, Zhang C, Ruan H, Yang B (2021) Thyroid hormone mediates cardioprotection against postinfarction remodeling and dysfunction through the IGF-1/PI3K/AKT signaling pathway. Life Sci 267:118977. https://doi.org/10.1016/j.lfs.2020.118977
Zeng B, Liu L, Liao X, Zhang C (2021) Cardiomyocyte protective effects of thyroid hormone during hypoxia/reoxygenation injury through activating of IGF-1-mediated PI3K/Akt signalling. J Cell Mol Med 25:3205–3215. https://doi.org/10.1111/jcmm.16389
Jianhua Z, Wilson GF, Soerens AG, Koonce CH, Junying Yu, Palecek SP, Thomson JA, Kamp TJ (2009) Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res 104:e30–e41. https://doi.org/10.1161/CIRCRESAHA.108.192237
Zhao D, Guallar E, Ouyang P, Subramanya V, Vaidya D, Ndumele CE, Lima JA, Allison MA, Shah SJ, Bertoni AG, Budoff MJ, Post WS, Michos ED (2018) Endogenous sex hormones and incident cardiovascular disease in post-menopausal women. J Am Coll Cardiol 71:2555–2566. https://doi.org/10.1016/j.jacc.2018.01.083
Zhao J, Pei L (2020) Cardiac endocrinology: heart-derived hormones in physiology and disease. JACC Basic Transl Sci 5:949–960. https://doi.org/10.1016/j.jacbts.2020.05.007
Zhou R, Li J, Zhang L, Cheng Y, Yan J, Sun Y, Wang J, Jiang H (2020) Role of Parkin-mediated mitophagy in glucocorticoid-induced cardiomyocyte maturation. Life Sci 255:117817. https://doi.org/10.1016/j.lfs.2020.117817
Ziman AP, Gómez-Viquez NL, Bloch RJ, Lederer WJ (2010) Excitation-contraction coupling changes during postnatal cardiac development. J Mol Cell Cardiol 48:379–386. https://doi.org/10.1016/j.yjmcc.2009.09.016
Acknowledgements
The authors acknowledge BioRender.com for providing the toolbox that has been used to create all figures.
Funding
Open Access funding enabled and organized by Projekt DEAL. No funding was received.
Author information
Authors and Affiliations
Contributions
The idea for the article originated from AH. The literature search was performed by AH and A-MG. The first draft of the manuscript was written by A-MGalowand critically revised by JB and AH. Anne-Marie Galow created the figures using Biorender.com. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Galow, AM., Brenmoehl, J. & Hoeflich, A. Synergistic effects of hormones on structural and functional maturation of cardiomyocytes and implications for heart regeneration. Cell. Mol. Life Sci. 80, 240 (2023). https://doi.org/10.1007/s00018-023-04894-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-04894-6