
Abstract
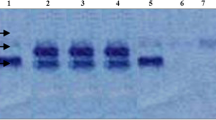
Mutations at the hexosaminidase A (HEXA) gene which cause Tay-Sachs disease (TSD) have elevated frequency in the Ashkenazi Jewish and French-Canadian populations. We report a novel TSD allele in the French-Canadian population associated with the infantile form of the disease. The mutation, a G→A transition at the +1 position of intron 7, abolishes the donor splice site. Cultured human fibroblasts from a compound heterozygote for this transition (and for a deletion mutation) produce no detectable HEXA mRNA. The intron 7+1 mutation occurs in the base adjacent to the site of the adult-onset TSD mutation (G805A). In both mutations a restriction site for the endonuclease EcoRII is abolished. Unambiguous diagnosis, therefore, requires allele-specific oligonucleotide hybridization to distinguish between these two mutant alleles. The intron 7+1 mutation has been detected in three unrelated families. Obligate heterozygotes for the intron 7+1 mutation were born in the Saguenay-Lac-St-Jean region of Quebec. The most recent ancestors common to obligate carriers of this mutation were from the Charlevoix region of the province of Quebec. This mutation thus has a different geographic centre of diffusion and is probably less common than the exon 1 deletion TSD mutation in French Canadians. Neither mutation has been detected in France, the ancestral homeland of French Canada.
Similar content being viewed by others

References
Akli S, Chelly J, Lacorte J-C, Poenaru L, Kahn A (1991) Seven novel Tay-Sachs mutations detected by chemical mismatch cleavage of PCR-amplified cDNA fragments. Genomics 11:124–134
Andermann E, Scriver CR, Wolfe LS, Dansky L, Andermann F (1977) Genetic variants of Tay-Sachs disease: Tay-Sachs disease in French Canadians, juvenile Tay-Sachs disease in Lebanese Canadians and a Tay-Sachs screening program in the French Canadian population. Prog Clin Biol Res 18:161–188
Arpaia E, Dumbrille-Ross A, Maler T, Neote K, Tropak M, Troxel C, Stirling JL, Pitts JS, Bapat B, Lambonwah AM, Mahuran DJ, Schuster SM, Clarke JTR, Lowden JA, Gravel RA (1988) Identification of an altered splice site in Ashkenazi Tay-Sachs disease. Nature 333:85–86
Charbonneau M, Robert H (1987) The French origin of the Canadian population 1608–1759. In: Harris RC (ed) Historical atlas of Canada, vol 1. University of Toronto Press, Toronto, p 118–119
De Braekeleer M, Morgan K, Jomphe M, Bouchard G, Davignon J, Gradie M, Kessling A, Laberge C, Moorjani S, Roy M, Scriver CR (1988) Familial hypercholesterolemia in French Canadians: geographical distribution and centre of origin of an LDL-receptor deletion mutation. (Technical report III-C-60) SOREP, Chicoutimi, Quebec
De Braekeleer M, John S, Leggett D, Laframboise R, Laberge C, Rozen R, Scriver CR (1990) A center of diffusion in 17th century France for the M1V allele in French Canadians. Am J Hum Genet 47:A131
De Braekeleer M, Hechtman P, Andermann E, Kaplan F (1992) The French-Canadian Tay-Sachs disease deletion mutation: identification of probable founders. Hum Genet 89:83–87
Ferre F, Garduno S (1989) Preparation of crude cell extract suitable for amplification of RNA by the polymerase chain reaction. Nucleic Acids Res 17:2141
Gauvreau D, Bourque M (1988) Mouvements migratoires et familles: le peuplement du Saguenay avant 1911. Rev Hist Am Fr 42:167–191
Grebner EE, Tomczak J (1991) Distribution of three alpha-chain beta-hexosaminidase A mutations among Tay-Sachs carriers. Am J Hum Genet 48:604–607
Hechtman P, Kaplan F, Bayleran J, Boulay B, Andermann E, De Braekeleer M, Melancon S, Lambert M, Potier M, Gagné R, Kolodny E, Clow C, Capua A, Prevost C, Scriver CR (1990) More than one mutant allele causes infantile Tay-Sachs disease in French Canadians. Am J Hum Genet 47:815–822
Jomphe M, Bouchard G, Davignon J, DeBraekeleer M, Gradie M, Kessling A, Laberge C, Moorjani S, Morgan K, Roy M, Scriver CR (1988) Familial hypercholesterolemia in French-Canadians: geographic distribution and center of origin of an LDL-receptor deletion mutation. Am J Hum Genet 43:A216
Kaplan F, Boulay B, Bayleran J, Hechtman P (1991) Allele-specific amplification of genomic DN A for detection of deletion mutations: identification of a French-Canadian Tay-Sachs mutation. J Inherited Metab Dis 14:707–714
Maniatis T, Fritsch EF, Sambrook J (1982) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory. Cold Spring Harbor, NY
McDowell GA, Fabacher P, Shapira E, Blitzer MG (1991) Characterization and distribution of Tay-Sachs disease alleles in the Cajun population. Am J Hum Genet 49 [suppl]:A2709
Myerowitz R (1988) A splice junction mutation in some Ashkenazi Jews with Tay-Sachs disease: evidence against a single defect within this ethnic group. Proc Natl Acad Sci USA 85:3955–3959
Myerowitz R, Costigan FC (1988) The major defect in Ashkenazi Jews with Tay-Sachs disease is an insertion in the gene for the alpha-chain of beta-hexosaminidase. J Biol Chem 263:18587–18589
Myerowitz R, Hogikyan ND (1986) Different mutations in Ashkenazi Jewish and non-Jewish French Canadians with Tay-Sachs disease. Science 232:1646–1648
Myerowitz R, Hogikyan ND (1987) A deletion involving Alu sequences in the beta-hexosaminidase alpha-chain gene of French Canadians with Tay-Sachs disease. J Biol Chem 262:15369–15399
Navon R, Kolodny EH, Mitsumoto H, Thomas GH, Proia RL (1990) Ashkenazi-Jewish and non-Jewish adult GM2 gangliosidosis patients share a common genetic defect. Am J Hum Genet 46:817–821
Ohno K, Suzuki K (1988) A splicing defect due to an exon-intron functional mutation results in abnormal beta-hexosaminidase alpha chain mRNAs in Ashkenazi Jewish patients with TaySachs disease. Biochem Biophys Res Commun 153:463–469
Orita M, Suzuki Y, Sekiya T, Hayashi K (1988) Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerase chain reaction. Genomics 5:874–879
Paw BH, Kaback MM, Neufeld EF (1989) Molecular basis of adult-onset and chronic GM2 gangliosidosis in patients of Ashkenazi Jewish origin: substitution of serine for glycine at position 269 of the alpha-subunit of beta-hexosaminidase. Proc Natl Acad 86:2413–2417
Paw BH, Tieu PT, Kaback MM, Lim J, Neufeld EF (1990) Frequency of three Hex A mutant alleles among Jewish and nonJewish carriers identified in a Tay-Sachs screening program. Am J Hum Genet 47:698–705
Triggs-Raine BL, Gravel RA (1990) Diagnostic heteroduplexes: simple detection of carriers of a 4-bp insertion mutation in Tay-Sachs disease. Am J Hum Genet 46:183–184
Triggs-Raine BL, Akermann BR, Clarke JTR, Gravel RA (1991) Sequence of DNA flanking the exons of the HEXA gene, and identification of mutations in Tay-Sachs disease. Am J Hum Genet 49:1041–1055
Triggs-Raine BL, Feigenbaum AS, Natowicz M, Skomorowski M-A, Schuster SM, Clarke JTR, Mahuran DJ, Kolodny EH, Gravel RA (1990) Screening for carrier of Tay-Sachs disease among Ashkenazi Jews: a comparison of DNA-based and enzymebased tests. N Engl J Med 323:6–12
Trop I, Kaplan F, Hechtman P, Brown C, Mahuran D (1992) A Glycine250→Aspartate substitution in the α subunit of hexosaminidase A causes juvenile-onset Tay-Sachs disease in a Lebanese-Canadian family. Hum Mut 1:35–39
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Hechtman, P., Boulay, B., De Braekeleer, M. et al. The intron 7 donor splice site transition: a second Tay-Sachs disease mutation in French Canada. Hum Genet 90, 402–406 (1992). https://doi.org/10.1007/BF00220467
Received:
Revised:
Issue Date:
DOI: https://doi.org/10.1007/BF00220467