
Abstract
Present work is aimed at preparing hybrid composites based on graphene oxide and hydroxyapatite as a matrix (GO/HA), which can be used as a template for drug delivery after chirurgical operation. Composite materials were synthesized through an in situ precipitation as simple and save-time method. The obtained materials were characterized using infrared spectroscopy (FT-IR), scanning electron microscopy (SEM), thermogravimetric analysis (TGA), and X-ray diffraction (XRD). Hybrid materials were loaded with amoxicillin and the drug delivery during various periods of time was assessed with UV spectroscopy. During loading periods, it was noted that GO has a much larger cumulative drug concentration compared to hydroxyapatite and other composites. Similarly, and during the release process, amoxicillin is released much faster during the earlier stage of immersion before reaching a stationary state towards long periods of time. This variation was found to be dependent on the amount of GO in the HA matrix, as well as on the number of oxygen available on the GO sheets and the HA surfaces.
Similar content being viewed by others

1 Introduction
The bone infection caused by microorganism on orthopaedic surgeries is the costliest complications, and is one of the major increased morbidity in the world [1]. This infection is associated with patient discomfort, longer hospitalization and additional surgeries for the treatment of infected patients [2,3,4,5]. Treatment of bone infection at surgical site is actually more demanded to prevent bacterial infections. Traditionally treatment of bone infection is ensured by administration of antibiotic systematically, but unfortunately, the drawbacks of this delivery route is the poor drug diffusion in the blood [6]. However, local delivery of antimicrobial drug on material as carrier, which is able to release the drug needed in the specific organ with a very defined concentration. Therefore, this is a viable solution to ensure a treatment of infection after orthopaedic surgeries [7].
Due to its fascinating properties, graphene has been accompanied by increasing research attention to explore this new material for energy conversion and storage systems [8, 9] to photocatalysis [10,11,12,13], through drug delivery application. Graphene (Gr), is a single layer at atomic thick, and a two- dimensional (2D) sp2 hybridized carbon material arranged in a honeycomb crystal lattice. Graphene had a wide variety of promising biomedical applications such as anti-bacterial paper, cancer targeting, photo-thermal therapy, drug delivery, biological imaging, and tissue engineering [14, 15]. Graphene has been widely used in his oxidized structure called graphene oxide (GO) due to the abundant hydroxyl, epoxide, and carboxylic groups on the basal plane and edges of graphene oxide surface respectively, which lead to the high dispersion of GO in water [16]. Consequently, makes their biomedical application in drug loading and delivery possible [17,18,19]. In addition the large specific surface area (2630 m2 g−1), the good biocompatibility, and high capability of loading drugs through physical adsorption, make GO an excellent carrier for loading and delivery for a variety of therapeutic drugs, such as doxorubicin, docetaxel, and camptothecin [20,21,22]. However, controlled drug release rate of GO is essentially very demanded for the performance of drug materials. In fact, if the drug release rate is faster, it can consequently kill some normal cells. In the other hand the drug release rate may not be slow, which can reduce their effective to treat bone infection [23].
Hydroxyapatite (HA) is actually an attractive biomaterial used in hard tissue engineering for restoring damaged bone, owing to its biological properties, and chemical composition similar to natural bone [24]. Also, HA have a great potential as drug delivery system. First and foremost, the geometrical structure of hydroxyapatite was found to have a vital role in adsorption of biomolecules through electrostatic attractions. The crystallographic structure of HA is formed by two horizontal layer of \({\text{PO}}_{4}^{3 - }\) tetrahedron, located at z = ¼ and z = ¾ planes, and positively charged Ca2+ atoms located at z = 0, ½, ¼ and ¾ planes and negatively charged OH− located at ¼, ¾ [25]. These two different charges of HA may be responsible for the adsorption of drugs and biomolecules on its surface. In addition, HA is very demanded for its potential application on drug delivery, thanks to the capacity to deliver anti-cancer drug [26], anti-inflammatory drug [27], anti-osteoporotic drug [28]. Therefore, the development of HA-drug systems, e.g. antibiotic, anti-microbial agent, etc., with controlled drug release seems to be an important problem, in view of the infection in bone surgery [29]. It is important to develop more advanced system able to deliver drug when required with optimal dosage within time.
Recently, few research works have introduced hydroxyapatite into graphene oxide in order to combine the advantages properties of GO and hydroxyapatite for drug delivery; and to develop composites with controlled drug release kinetics. For example, pH-sensitive drug carriers have been investigated using GO/HA composites and the release rate from these composites was found to be pH-dependent. The drug release in PBS medium (phosphate buffer solution) at a pH = 4.4 was found to be higher compared to pH = 7.4 and 9 respectively [30]. Furthermore, the porous flower like structure of the GO/HA composites revealed a reduction in drug release rate of ibuprofen (IBU), which make this nanocomposite, a promising material for drug delivery application [31]. Also, drug release of a cationic Anticancer doxorubicin was controlled on carbon dots conjugated carboxymethyl cellulose-hydroxyapatite nanocomposite which explained by a diffusion-controlled mechanism [32]. Even more, there is a serious discrepancy about the future application of hydroxyapatite particles, when they are combined with GO, if the combination will be able to control drug release or not. The big challenge must be achieved is the new composites development functionalized by therapeutic drug to significantly improve local surgical outcomes and reduced bone infection. Despite the progress in graphene based nano-composites, to our knowledge, there are few reports on GO/HA composites as a template for drug delivery to control release drug rate. In this paper, the effect of GO loading and drugs release will be studied. The drug loading and controlled release will be investigated using Amoxicillin (Fig. 1) as the model drug.

Chemical structure of Amoxicillin drug molecule
2 Experimental details
2.1 Materials
Synthetic Graphite (powder, ˂ 20 mm), amoxicillin, sulfuric acid (H2SO4), sodium nitrate (NaNO3), potassium permanganate (KMnO4), hydrogen peroxide (H2O2), chloridric acid (HCl), calcium chloride (CaCl2), ammonia water (NH4OH), phosphoric acid (H3PO4) were purchased from Sigma Aldrich Chemicals. All the chemicals were employed without further purification.
2.2 Apparatus and characterization
Fourier transform infrared (FTIR) analyses were carried out on a Thermoscientific, IS-50 FT-IR in the frequency range of 4000–400 cm−1 to identify the functional groups of the composites, with a resolution of 4 cm−1. The X-ray diffraction (XRD) patterns of the powders and composites were assayed using an automated X-ray powder diffractometer (XRD, PAnalytical) at a scanning rate of 0.033° per second in a 2θ range from 20° to 80° with Cu-Kα radiation (λ = 1.54,060 Å), operated at 45 kV and 40 mA. Thermogravimetric analysis (TGA) was conducted with TGA Q500. All the samples were carefully grounded to fine powder. The samples were analyzed within the temperature range 25–1000 °C at a heating rate of 10 °C/min under atmospheric environment. The morphology of the hybrids was carried out by using scanning electron microscopy (SEM) on FEI Quanta 200 EDAXR.
2.3 Synthesis of graphene oxide composites (GO)
Graphene oxide (GO) was synthesized using Hummer’s method from natural graphite. Briefly, 2 g of graphite powder and 1 g of NaNO3 were added to 50 mL of concentrated H2SO4 and were stirred in an ice bath, then 6 g of KMnO4 was slowly added with vigorous stirring. The suspension was kept at 35 °C for 30 min with stirring. After that, the reaction was heated at 95 °C and stirred for 30 min after addition of 180 ml of water. Then, 450 ml of water was slowly added following by the addition of 15 ml H2O2 (30%) slowly. The mixture was filtered and washed with HCl (1 M, 37%) and then with distilled water, until the suspension was neutral and dried at 60 °C for 24 h [33].
2.4 In situ synthesis of HA in graphene oxide suspension
Predetermined amount of graphene oxide was dispersed by ultra-sonication in 50 ml of water for 2H. Hydroxyapatite was prepared in the presence of GO solution at different weight fraction of GO by in situ precipitation (Table 1).
CaCl2 precursors (10.25 mM) were dissolved in 50 ml of water and the solution was added drop wise to GO solution under stirring. After that, 50 ml of diluted H3PO4 (6.14 mM) was added to the previous solution under agitation during 30 min. The pH of the solution was then adjusted to 10 by adding ammonia solution (NH4OH). The solution was then stirred for 24 h at 37 °C. the resulting precipitate was centrifuged and washed four times with distilled water and ethanol. The obtained powder was dried at 80 °C for overnight.
2.5 In vitro tests: drug loading, release and biomimetic mineralization
Loading of amoxicillin (AMX) was carried out by dispersing GO/HA (a, b, c, d, e) in aqueous solution at pH = 7.42 at 37 °C for 24H in dark environment. The liquid supernatant was separated from solid samples using centrifugation operating at 10000 rpm for 5 min. The drug loading was calculated according to the Eq. (3). The release of AMX from various composite samples (GO/HA) was carried out by dispersing pre-weighed samples in PBS medium at pH = 7.42, T = 37 °C (in an incubator) at various period of times (1, 2, 4.5, 5.5, 24, 48, 72 and 96H). Periodically, 4 ml of dissolution medium were collected and replaced by the same volume. The PBS solutions were analysed at 272 nm by UV/VIS Spectrometer Lamda-850. The experiments (loading and release) were repeated in triplicate to get mean and the standard deviation. The amount of the AMX released was calculated using a linear regression. The cumulative release of AMX was calculated from Eq. (2):
m0 is the initial mass of drug used in loading (1 mg/ml), mt is the mass of AMX collected in aqueous or PBS solution, t is the time of collection.
To investigate the apatite forming ability, cylindrical shaped samples weighing 0.1 g prepared by uniaxial press were soaked in 30 mL of SBF [34] solution in a polyethylene plastic container and were placed in an incubator at 37 °C (± 0.5 °C). After 21 days of immersion, the samples were collected from the SBF solution, rinsed with water and dried at room temperature. SEM observation was used to check the apatite formation on the surfaces of cylindrical shaped samples.
3 Results and discussion
3.1 Preparation and characterization of GO/HA composites
Scheme 1 illustrates the synthesis process of the composites GO/HA (a, b, c, d, and e). The hybrid materials were prepared using in situ precipitation method. First, GO was ultrasonically well dispersed in water. Then, in situ precipitation of HA was achieved. During the reaction process, polar functional groups of GO surface act as nucleation and growth sites of HA. First, calcium ions (Ca2+) with positive charge was strongly attracted to GO surfaces via electrostatic interaction. Then, phosphate ions (\({\text{PO}}_{4}^{3 - }\)) will counterbalance the electronic charge of the surface. The increase of the pH of the solution to 10–10.5, will spontaneously induced nucleation and growth of HA on GO surfaces within time.

The experimental process of synthesized composites
3.2 FTIR and UV analysis of GO and GO/HA composites
FTIR spectra of GO and GO/HA (a, b, c, d, e) composites are displayed in Fig. 2. According to Fig. 2a, the absorbance bands at 3203 cm−1 and 1246 cm−1 were assigned to the hydroxyl groups (O–H), 1620 cm−1 to the sp2-hybridized C=C vibration stretching. Besides, the adsorption peaks produced by other functional groups such as carboxyl C=O (1730 cm−1), epoxy C–O (1051 cm−1) groups on the surface of GO [35]. After nucleation and growth of HA particles, the future composites were examined with FTIR. Infrared spectra of GO/HA composites showed all characteristic peaks of hydroxyapatite at 560, 600,967 and 3345 cm−1 respectively (Fig. 2b) [36]. Moreover, according Fig. 2b, it was observed a net reduction of pic intensity of the absorption band of C=O (1730 cm−1) indicating a strong interaction between HA and GO nanosheets. It was also observed from UV–vis spectra of GO and GO/HA composite (Fig. 2d) that GO nanosheets consist of the band at 232 nm, which is characteristic of π–π* transitions of carboxyl group (C=O) [37]. After addition of HA, the peak located at 232 nm was shifted to 310 nm, which confirmed the strong interaction between HA and GO [31, 38].

FTIR spectra of a graphite and graphene oxide, b GO/HA corresponding composites and c UV–vis spectra of graphene oxide (GO) and selected GO/HA composite
3.3 X-ray diffraction studies and thermogravimetric analysis
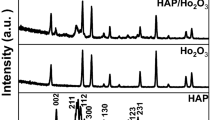
Phase and crystallinity of prepared composites were identified using XRD analysis. Figure 3a shows the XRD pattern of graphite and graphene oxide where graphite shows an intense peak at 2θ = 26.5° and GO at 2θ = 10.1° (due to (002) plane) which clearly indicates the oxidation of graphite affecting its crystal structure and the interlayer spacing which has increased from 3.4 to 8.7 Å [35]. Figure 3b displayed the XRD patterns of the GO/HA composites and HA powders. All samples presented diffraction peaks of HA crystal at 2θ of 25.79, 31.9, 39.3, 46.36, 49.5, 53.26 corresponding to (002), (211), (310), (222), (313), (004) inter-reticular plans [39]. These characteristic peaks are consistent with the standard diffraction pattern of HA (JCPDF 00-001-1008). However, after formation of HA on GO sheets, the (002) plan of GO was not observed in the composites, due the lower amount of GO, which was consistent with previous studies [40].

a XRD patterns of Graphite and GO, b XRD patterns of GO/HA composite materials
TGA analysis were further performed to confirm the weight change of GO, HA, and GO/HA (a, b, c, d, e) (Fig. 4). The samples were heated from room temperature to 1000 °C at a rate of 10 °C/min. The initial weight loss at around 100 °C was attributed to the evaporation of water for all samples. According to this figure, the GO is thermally unstable and recorded two significant loss weight for at around 200 °C and 510 °C due to decomposition of oxygenated functional groups. The former are attributed to formation of CO, and CO2, and the latter are attributed to the combustion of the carbon of GO [41]. The weight loss of GO at 200 °C was 20 wt%, compared to GO/HA (a) to GO/HA (e) that show a variation of weight loss from 2.5 to 4.5 wt% respectively. In addition, when the temperature reached 900 °C, no significant weight loss was recorded for HA, and GO/HA composites (a, b, c, d, and e), which due to their high thermal stability of HA. The final weight loss was also found to be GO dependent. the higher the graphene loading rate in composite materials, the more a net decrease in mass loss is recorded at 900 °C.

TGA profiles of HA, GO and GO composite
3.4 AMX loading and in vitro drug release
According to FTIR spectra of AMX (Fig. 5), the absorbance bands at 1770 cm−1, 1640–1560 cm−1, 1350–1280 cm−1, 3650–3590 cm−1 were corresponded to ketone, primary amine, secondary amine, and hydroxyl (broad) groups of amoxicillin respectively [42]. The characteristic bands of AMX were also observed for all composite materials (GO/HA) which confirmed the successful loading of AMX drug molecule in all samples. As mentioned above, the UV–Vis spectroscopy displayed a characteristic absorbance band at 272 nm for amoxicillin (Fig. 2c). The loading content in composite materials were assessed by UV spectroscopy and for that the calibration curve for various concentration of AMX was released.

FTIR spectra of GO/HA composite samples loaded with AMX drug molecule
Figure 6a shows the evolution of the concentration of AMX in GO, HA and their corresponding composites. With respect to this figure, it can be observed that virgin graphene exhibited the highest absorbance of the AMX drug molecules (82.19%) compared to HA (51.99%) and the other composites. This can be explained by the abundance of the graphene-sheets by functional groups capable of developing favourable interactions (π–π interactions) with AMX such as carboxyl, epoxide and carboxylic acid [43]. The second parameter impacting this extraordinary absorbance of the AMX drug molecules is the higher specific surface area of graphene sheets.

AMX loading (a) and release (b) profiles from HA, GO and GO/HA carriers
In the case of HA, the average cumulative concentration of the AMX absorbed on the surface of the granules reached 51.99%. This modest value compared to that obtained in the case of the GO, is explained by the methodology adopted during synthetizing the HA. It should be noted that the preparation of the HA by the precipitation method with vigorous stirring does not adequately allowed a good control of the geometry of the grains, neither it can increase the specific surface area of the apatite particles such as the case observed whit surfactant assisted particle synthesis of hydroxyapatite [29]. This will effectively reduce the exposure of functional groups able of developing favourable interactions with AMX.
For the intermediate phases, it has been observed that the cumulative concentration of AMX increased with GO addition in the prepared composites. In the case of GO/HA composites (a, 0.5 wt% of graphene), a value of 45.42% was recorded. This value is slightly lower to that observed in the case of the virgin HA and can be explained by a net reduction of the oxygenated sites necessary to interact with the AMX drug molecules. Generally, during the formation of hydroxyapatite in the presence of suspended graphene sheets, the formation of covalent bonds between HA and GO has been observed and may be responsible for this decrease. This has been also confirmed by the FTIR spectra (Fig. 2b). As the amount of GO increased in the prepared composites, the cumulative concentration of AMX increase to reach its higher value in the case of 3 wt% of GO addition. The available of both functional groups on nanosheets of GO and HA are responsible of this enhancement. In addition, it was reported earlier that the percentage of graphene filled HA should not exceed 5 wt% to not deteriorate biological cells [44,45,46].
Figure 6b shows the kinetics of release of AMX drug molecules in PBS medium for virgin materials (HA and GO) and their corresponding composites. According to this figure, it can be seen that the drug release is graphene oxide dependent. As a function of time, all composites presented a net increase in the AMX concentration in the PBS solution during the earlier stage of immersion (for periods of time less than 5 h) before reaching a quasi-stationary states after 5 h of immersion. The steady states are therefore dependent on the amount of graphene in composite materials. As the rate of graphene oxide was increased in the composite materials, as the concentration limit of AMX in the PBS solution become more important. Graphene oxide therefore has the highest concentration while HA has the lowest one. This phenomenon is explained by the amount of AMX absorbed on the surface of composites during drug loading as well as by the strength of the concentration gradient during the release in the PBS solution. The rapid delivery of the drug during the earlier stage is required immediately after surgery for the effective inhibition of microorganisms and then a controlled release is needed to aid long-time healing and to avoid the toxic and adverse systemic effect caused by high concentration of antibiotics administrated orally.
In order to qualify the mechanism of AMX release, several researchers suppose that drug release mechanism occurred by either drug diffusion or eroding of the matrix, which influenced the rate of release. Several models have been used to explain the mechanisms governing the drug release. Semi-empirical models were developed by Ritger & Peppas (Eq. 3) [47] and kopcha (Eq. 4) [48,49,50].
\(\frac{{{\text{M}}_{\text{t}} }}{{{\text{M}}_{\infty } }}\) is the fractional solute release, Mt is the amount of released drug at time t, M∞ is total amount of released drug, k is a constant, and n is the diffusional exponent characteristic of the release mechanism. n gives an information of the drug-release mechanism: if n < 0.5, the mechanism corresponded to Fickian diffusion and if n is situated between 0.5 and 1.0, the mechanism will correspond to non-Fickian transport. For Eq. 4, A is a diffusional term and B is the erosion term. When A/B > 1, the diffusion phenomenon is predominant, and when A/B < 1 the erosion phenomenon is predominant. Table 2 represents the results of calculations of parameters n, A and B respectively. According to the calculated parameters, it can be concluded from both models, that the drug release followed the Fickian diffusion law for all samples.
3.5 Biomimetic mineralization
Two factors are consistent from the point of view of good implants grafting: good osseointegration of the biomaterials with surrounding tissues and excellent biocompatibility of the biomaterials for the growth promotion of osteoblast cells [51]. A strong bone-bonding ability between the implant and the surrounding tissues is generally due to a regular distribution and fast apatite formation. In this study, we used in vitro acellular bioactivity tests using simulated body fluid (SBF) as a standard precursor medium mimicking the biomineralization process of the new bone. In order to evaluate this capacity, GO/HA composite materials were immersed in SBF for different periods of time (1–21 days). All GO/HA composite samples showed the formation of a new apatite layers as consistently confirmed by SEM and FTIR results. Figure 7 displayed the representative surface morphologies obtained from SEM of pure HA and the GO/HA composites after being soaked in SBF for 21 days.

SEM micrographs of a GO sheets, b HA before soaking, c HA, d GO/HA (0.5%), e GO/HA (2%), f GO/HA (3%) after 21 days of soaking in SBF medium
Figure 7a revealed that GO appears like folded flakes, multi layered and wavy, these observations are consistent with those from Mohandes et al. [52] study. However, HA pellet displayed a smooth surface before soaking in SBF medium (Fig. 7b). In contrary, it’s clearly observed that the morphology of mineralization product varies with the addition of GO into the HA matrix and varied from spherical to sheet-like apatite forms. Increasing the amount of GO in the composites promote the deposition of a dense layer of spherical-like apatite, although pure HA ceramics showed a platelet-like crystals with the typical HA morphology. In addition, it is observed some microcraks on the GO containing samples, indicating the formation of a thick deposit [53]. The apatite layer deposition is controlled by the presence of available oxygen functionalities on GO and HA surfaces. Thereafter, both of them proceed as competing nucleation points for ionic cluster precursors. For pure HA, the same situation is observed in the case of the formation of apatite on the surface of sintered ceramics after soaking in SBF, which allows for selective ion diffusion [54,55,56].
In the case of GO/HA composite materials, the formation of apatite layer after 21 days of immersion (Fig. 7d–f). They specifically grow on the HA particles, and interestingly, few of them appear on divers functional groups present on GO nanosheets, which remain accessible after the in situ synthesis of hydroxyapatite. These observations suggested that HA nuclei forming on the GO/HA surface retain a high reactivity which allows them to migrate towards the GO/HA surface to initiate HA growth and resulted on a dense apatite layer. All the observations showed clearly that the presence of hydroxyapatite and GO nanosheets are recommended to obtain a rough surface after the deposition of the new apatite layers.
As known, that this rough surface is ideal for the better adhesion of cells [57]. The formation of apatite particles on the various hybrid composites surfaces is followed more closely by FTIR spectroscopy (Fig. 8). After incubation for 21 days, the FTIR pattern revealed the presence of \({\text{PO}}_{4}^{3 - }\) and OH groups. Weak \({\text{PO}}_{4}^{3 - }\) bands appeared for HA samples. After further addition of GO, strong \({\text{PO}}_{4}^{3 - }\) bands were observed. Among these bands, the band at 964 cm−1 is due to the symmetric stretching mode ν1 (\({\text{PO}}_{4}^{3 - }\)), 1015 and 1089 cm−1 to the vibration mode ν3 (\({\text{PO}}_{4}^{3 - }\)), 597, 560 cm−1 and 472 cm−1 to bending modes ν4 (\({\text{PO}}_{4}^{3 - }\)). Then, the FTIR results also confirmed the formation of apatite phase on the GO/HA composites surfaces after soaking in SBF medium [36, 58]. The most significant and interesting finding is that hydroxyapatite and graphene oxide are a promising combination for bone implant applications [57].

FTIR spectra of the pure HA as reference, and pure HA, GO/HA (a, d, e) after 21 days of soaking in SBF medium
4 Conclusions
To summarize, composite materials based on hydroxyapatite and graphene oxide were synthetized through in situ precipitation as simple and save-time method. Hybrid materials were loaded with amoxicillin as drug molecule and drug release was assessed by UV–Vis spectroscopy. In addition, the drug release was found to be GO dependent and the kinetics of drug release had a tendency to fit well with Fickian diffusion model. Moreover, the composite materials sustained the release of phosphate and calcium ions in SBF, which synergistically assists the ECM (Extracellular matrix) ossification and promotes new bone regeneration. Furthermore, the GO contentment was found to accelerate the process of biomineralization and promoted new bone formation.
References
Noukrati H, Cazalbou S, Demnati I et al (2016) Injectability, microstructure and release properties of sodium fusidate-loaded apatitic cement as a local drug-delivery system. Mater Sci Eng C 59:177–184. https://doi.org/10.1016/j.msec.2015.09.070
Green JW, Wenzel RP (1977) Postoperative wound infection: a controlled study of the increased duration of hospital stay and direct cost of hospitalization. Ann Surg 185:264–268. https://doi.org/10.1097/00000658-197703000-00002
Kluytmans JAJW, Mouton JW, Maat APWM et al (1994) Surveillance of postoperative infections in thoracic surgery. J Hosp Infect 27:139–147. https://doi.org/10.1016/0195-6701(94)90007-8
Kluytmans J, Mouton JW, Ijzerman EPF et al (1995) Nasal carriage of Staphylococcus aureus as a major risk factor for wound infections after cardiac surgery. J Infect Dis 171:216–219
Coellot R, Glenister H, Fererest J et al (1993) The cost of infection in surgical patients: a case-control study. J Hosp Infect 25:239–250
Dubnika A, Loca D, Rudovica V et al (2017) Functionalized silver doped hydroxyapatite scaffolds for controlled simultaneous silver ion and drug delivery. Ceram Int 43:3698–3705. https://doi.org/10.1016/j.ceramint.2016.11.214
Reina G, González-Domínguez JM, Criado A et al (2017) Promises, facts and challenges for graphene in biomedical applications. Chem Soc Rev 46:4400–4416. https://doi.org/10.1039/c7cs00363c
Hughes G, Trigg J (2009) Remembering the charge of the light brigade: its commemoration, war memorials and memory. Bastions Barbed Wire Stud Archaeol Confl. https://doi.org/10.1002/anie.201806221
Quan Q, Lin X, Zhang N, Xu YJ (2017) Graphene and its derivatives as versatile templates for materials synthesis and functional applications. Nanoscale 9:2398–2416. https://doi.org/10.1039/c6nr09439b
Lu KQ, Xin X, Zhang N et al (2018) Photoredox catalysis over graphene aerogel-supported composites. J Mater Chem A 6:4590–4604. https://doi.org/10.1039/c8ta00728d
Zhang N, Yang MQ, Liu S et al (2015) Waltzing with the versatile platform of graphene to synthesize composite photocatalysts. Chem Rev 115:10307–10377. https://doi.org/10.1021/acs.chemrev.5b00267
Han C, Zhang N, Xu YJ (2016) Structural diversity of graphene materials and their multifarious roles in heterogeneous photocatalysis. Nano Today 11:351–372. https://doi.org/10.1016/j.nantod.2016.05.008
Yang MQ, Zhang N, Pagliaro M, Xu YJ (2014) Artificial photosynthesis over graphene-semiconductor composites. Are we getting better? Chem Soc Rev 43:8240–8254. https://doi.org/10.1039/c4cs00213j
Zhang Y, Nayak TR, Hong H, Cai W (2012) Graphene: a versatile nanoplatform for biomedical applications. Nanoscale 4:3833–3842. https://doi.org/10.1039/c2nr31040f
Wu X, Zheng S, Ye Y et al (2018) Enhanced osteogenic differentiation and bone regeneration of poly(lactic-: Co -glycolic acid) by graphene via activation of PI3K/Akt/GSK-3β/β-catenin signal circuit. Biomater Sci 6:1147–1158. https://doi.org/10.1039/c8bm00127h
Depan D, Pesacreta TC, Misra RDK (2014) The synergistic effect of a hybrid graphene oxide-chitosan system and biomimetic mineralization on osteoblast functions. Biomater Sci 2:264–274. https://doi.org/10.1039/c3bm60192g
Sun X, Liu Z, Welsher K et al (2008) Nano-graphene oxide for cellular imaging and drug delivery. Nano Res 1:203–212. https://doi.org/10.1007/s12274-008-8021-8
Liu Z, Robinson JT, Sun X, Dai H (2008) PEGylated nano-graphene oxide for delivery of water insoluble cancer drugs (b). J Am Chem Soc 130:10876–10877. https://doi.org/10.1021/ja803688x
Yang X, Zhang X, Ma Y et al (2009) Superparamagnetic graphene oxide–Fe3O4 nanoparticles hybrid for controlled targeted drug carriers. J Mater Chem 19:2710. https://doi.org/10.1039/b821416f
Zhao X, Liu P (2014) Biocompatible graphene oxide as a folate receptor-targeting drug delivery system for the controlled release of anti-cancer drugs. RSC Adv 4:24232–24239. https://doi.org/10.1039/C4RA02466D
Liu J, Cui L, Losic D (2013) Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater 9:9243–9257. https://doi.org/10.1016/j.actbio.2013.08.016
Yang Y, Wang Y, Zhu M et al (2017) RGO/AuNR/HA-5FU nanocomposite with multi-stage release behavior and efficient antitumor activity for synergistic therapy. Biomater Sci 5:990–1000. https://doi.org/10.1039/c7bm00007c
Wang H, Sun D, Zhao N et al (2014) Thermo-sensitive graphene oxide-polymer nanoparticle hybrids: synthesis, characterization, biocompatibility and drug delivery. J Mater Chem B 2:1362–1370. https://doi.org/10.1039/c3tb21538e
Rasouli R, Barhoum A, Uludag H (2018) A review of nanostructured surfaces and materials for dental implants: surface coating, patterning and functionalization for improved performance. Biomater Sci 6:1312–1338. https://doi.org/10.1039/c8bm00021b
Cazalbou S, Combes C, Eichert D, Rey C (2004) Adaptative physico-chemistry of bio-related calcium phosphates. J Mater Chem 14:2148–2153
Xiong H, Du S, Ni J et al (2016) Mitochondria and nuclei dual-targeted heterogeneous hydroxyapatite nanoparticles for enhancing therapeutic efficacy of doxorubicin. Biomaterials 94:70–83. https://doi.org/10.1016/j.biomaterials.2016.04.004
Öner M, Yetiz E, Ay E, Uysal U (2011) Ibuprofen release from porous hydroxyapatite tablets. Ceram Int 37:2117–2125. https://doi.org/10.1016/j.ceramint.2011.02.021
Seshima H, Yoshinari M, Takemoto S et al (2006) Control of bisphosphonate release using hydroxyapatite granules. J Biomed Mater Res Part B Appl Biomater 78B:215–221. https://doi.org/10.1002/jbm.b.30446
Komlev VS, Barinov SM, Koplik EV (2002) A method to fabricate porous spherical hydroxyapatite granules intended for time-controlled drug release. Biomaterials 23:3449–3454. https://doi.org/10.1016/S0142-9612(02)00049-2
Bharath G, Latha BS, Alsharaeh EH et al (2017) Enhanced hydroxyapatite nanorods formation on graphene oxide nanocomposite as a potential candidate for protein adsorption, pH controlled release and an effective drug delivery platform for cancer therapy. Anal Methods 9:240–252. https://doi.org/10.1039/c6ay02348g
Yao C, Zhu J, Xie A et al (2017) Graphene oxide and creatine phosphate disodium dual template-directed synthesis of GO/hydroxyapatite and its application in drug delivery. Mater Sci Eng C 73:709–715. https://doi.org/10.1016/j.msec.2016.11.083
Sarkar C, Chowdhuri AR, Kumar A et al (2018) One pot synthesis of carbon dots decorated carboxymethyl cellulose- hydroxyapatite nanocomposite for drug delivery, tissue engineering and Fe3 + ion sensing. Carbohydr Polym 181:710–718. https://doi.org/10.1016/j.carbpol.2017.11.091
Núñez JD, Benito AM, González R, Aragón A, Arenal R, Maser WK (2014) Integration and bioactivity of hydroxyapatite grown on carbon nanotubes and graphene oxide. Carbon 79:590–604
Kokubo T, Takadama H (2006) How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 27:2907–2915. https://doi.org/10.1016/j.biomaterials.2006.01.017
Li M, Wang Y, Liu Q et al (2013) In situ synthesis and biocompatibility of nano hydroxyapatite on pristine and chitosan functionalized graphene oxide. J Mater Chem B 1:475–484. https://doi.org/10.1039/c2tb00053a
Zeng Y, Pei X, Yang S et al (2016) Graphene oxide/hydroxyapatite composite coatings fabricated by electrochemical deposition. Surf Coat Technol 286:72–79. https://doi.org/10.1016/j.surfcoat.2015.12.013
Zhang J, Yang H, Shen G et al (2010) Reduction of graphene oxide via < scp > l</scp > -ascorbic acid. Chem Commun 46:1112–1114. https://doi.org/10.1039/B917705A
Shen J, Shi M, Yan B et al (2010) Covalent attaching protein to graphene oxide via diimide-activated amidation. Colloids Surf B Biointerfaces 81:434–438. https://doi.org/10.1016/j.colsurfb.2010.07.035
Butun S, Ince FG, Erdugan H, Sahiner N (2011) One-step fabrication of biocompatible carboxymethyl cellulose polymeric particles for drug delivery systems. Carbohydr Polym 86:636–643. https://doi.org/10.1016/j.carbpol.2011.05.001
Liu H, Xi P, Xie G et al (2012) Simultaneous reduction and surface functionalization of graphene oxide for hydroxyapatite mineralization. J Phys Chem C. https://doi.org/10.1021/jp2102226
Bissessur R, Liu PKY, White W, Scully SF (2006) Encapsulation of polyanilines into graphite oxide. Langmuir 22:1729–1734. https://doi.org/10.1021/la0527339
Patel DM, Patel DK, Patel CN (2011) Formulation and evaluation of floating oral in situ gelling system of amoxicillin. ISRN Pharm 2011:1–8. https://doi.org/10.5402/2011/276250
Bao H, Li L, Gan LH et al (2010) Thermo-and pH-responsive association behavior of dual hydrophilic graft chitosan terpolymer synthesized via ATRP and click chemistry. Macromolecules 43:5679–5687. https://doi.org/10.1021/ma100894p
Baradaran S, Moghaddam E, Basirun WJ et al (2014) Mechanical properties and biomedical applications of a nanotube hydroxyapatite-reduced graphene oxide composite. Carbon N Y 69:32–45. https://doi.org/10.1016/j.carbon.2013.11.054
Baradaran S, Moghaddam E, Nasiri-Tabrizi B et al (2015) Characterization of nickel-doped biphasic calcium phosphate/graphene nanoplatelet composites for biomedical application. Mater Sci Eng C 49:656–668. https://doi.org/10.1016/j.msec.2015.01.050
Crisan L, Crisan B, Soritau O et al (2015) In vitro study of biocompatibility of a graphene composite with gold nanoparticles and hydroxyapatite on human osteoblasts. J Appl Toxicol 35:1200–1210. https://doi.org/10.1002/jat.3152
Ritger PL, Peppas NA (1987) A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release 5:23–36. https://doi.org/10.1016/0168-3659(87)90034-4
Fan Z, Wang J, Wang Z et al (2013) Casein phosphopeptide-biofunctionalized graphene biocomposite for hydroxyapatite biomimetic mineralization. J Phys Chem C 117:10375–10382. https://doi.org/10.1021/jp312163m
Korsmeyer RW, Gurny R, Doelker E et al (1983) Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm 15:25–35. https://doi.org/10.1016/0378-5173(83)90064-9
Kopcha M, Lordi NG, Tojo KJ (1991) Evaluation of release from selected thermosoftening vehicles. J Pharm Pharmacol 43:382–387. https://doi.org/10.1111/j.2042-7158.1991.tb03493.x
Mehrali M, Moghaddam E, Shirazi SFS et al (2014) Mechanical and in vitro biological performance of graphene nanoplatelets reinforced calcium silicate composite. PLoS ONE. https://doi.org/10.1371/journal.pone.0106802
Mohandes F, Salavati-Niasari M (2014) Freeze-drying synthesis, characterization and in vitro bioactivity of chitosan/graphene oxide/hydroxyapatite nanocomposite. RSC Adv 4:25993–26001. https://doi.org/10.1039/c4ra03534h
Zhong H, Wang L, Fan Y et al (2011) Mechanical properties and bioactivity of β-Ca2SiO4ceramics synthesized by spark plasma sintering. Ceram Int 37:2459–2465. https://doi.org/10.1016/j.ceramint.2011.03.037
Kim HM, Himeno T, Kokubo T, Nakamura T (2005) Process and kinetics of bonelike apatite formation on sintered hydroxyapatite in a simulated body fluid. Biomaterials 26:4366–4373. https://doi.org/10.1016/j.biomaterials.2004.11.022
Belmamouni Y, Bricha M, Essassi EM, El Mabrouk K (2014) Fostering the mechanical strength and biomineralisation of nano-hydroxyapatite/multiwall carbon nanotubes (n-HA/MWCNTs) modified silane nanocomposite. J Biomater Tissue Eng 4:349–357. https://doi.org/10.1166/jbt.2014.1183
Bricha M, Belmamouni Y, Essassi EM et al (2013) Hydrothermal synthesis and appraisal of Mg-doped hydroxyapatite nanopowders. J Biomater Tissue Eng 3:570–580. https://doi.org/10.1166/jbt.2013.1120
Oyefusi A, Olanipekun O, Neelgund GM et al (2014) Hydroxyapatite grafted carbon nanotubes and graphene nanosheets: promising bone implant materials. Spectrochim Acta Part A Mol Biomol Spectrosc. https://doi.org/10.1016/j.saa.2014.04.004
Peng H, Meng L, Niu L, Lu Q (2012) Simultaneous reduction and surface functionalization of graphene oxide by natural cellulose with the assistance of the ionic liquid. J Phys Chem C 116:16294–16299. https://doi.org/10.1021/jp3043889
Acknowledgements
The Authors are grateful to the financial support allowable from Euromed University of Fes - Morocco and National Center for Scientific and Technical Research – Morocco, PPR2 Programme.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Taghzouti, O.K., El Mabrouk, K., Bricha, M. et al. Controlled adsorption and release of amoxicillin in GO/HA composite materials. SN Appl. Sci. 1, 226 (2019). https://doi.org/10.1007/s42452-019-0240-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-019-0240-y