
Abstract
In the present study we performed the comparative modeling, structural annotation, domain identification and the structural comparison of β-galactosidase enzyme from Aspergillus niger. Five domains were identified in the modeled structure at different residue regions. Two catalytic residues Glu200 and Glu298 were identified in the first domain of the modeled structure. The modeled structure showed highest similarity with crystal structure of Penicillium sp. β-galactosidase. The molecular docking of the structure with β-d-galactose was also performed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
β-Galactosidase (S-d-galactosides galactohydrolase, EC 3.2.1.23), is the enzyme responsible for the catalytic hydrolysis of β-galactosides into monosaccharides. The enzyme is an exoglycosidase which hydrolyzes the β-glycosidic bond formed between a galactose and its organic moiety. The enzyme deficiencies in the human body result in galactosialidosis or Morquio B syndrome. The enzyme gene, lacZ in Escherichia coli is present as an integral part of the inducing lac operon system and is positively regulated by the incidences of low lactose levels in the organism (Dorland 1997).
The functionality of the enzyme relies on the two structural peptides, lacZα and lacZΩ, neither of which is active by itself, but which are duly operative when coexisting in an aggregated entity. The presence or absence of an active β-galactosidase may be detected by X-gal, which produces a characteristic blue dye when cleaved by β-galactosidase, thereby providing an easy means of distinguishing the presence or absence of cloned product in a plasmid (Dimri et al. 1995).
The exclusive nature of the version of the enzyme found in Aspergillus niger is its thermostability at a wide tolerance range of 35–80 οC (Dimitris et al. 2005). The enzyme has significant role in A. niger in colony establishment, since it aids in breaking down complex sugars that can be readily assimilated by the fungi. Also, in large-scale industrial processes the fungi has been readily utilized for commercial production of β-galactosidase. The enzyme extracted from A. niger is useful in the commercial production of an array of sugars; glucose, galactose, heteropolysaccharides, galacto-oligosaccharides. β-Galactosidase based medical and industrial applications include cleavage of blood group A and B glycotypes, biosensor for specific lactose determination in milk and disease diagnosis (Staiano et al. 2005), treatment of lactose malabsorption, production of lactose hydrolyzed milk (Mlichová and Rosenberg 2006). Immobilization of β-galactosidase through anion exchange resin, cellulose–gelatin carrier system, DEAE agarose, glyoxyl/epoxy/BrCN groups, glutaraldehyde, polyelectrolyte surfaces, silicon surface, sepabeads-epoxy supports partially modified with boronate, iminodiacetic, metal chelates, and ethylenediamine improves its stability and re-usage (Klich 2002).
Considering the above facts, a greater insight into the properties of the β-galactosidase enzyme from A. niger would be more helpful in the working spectrum. In present study the comparative modeling, structural annotation, domain identification, and structural comparison of β-galactosidase enzyme from A. niger were performed. Further, the docking studies were carried out for catalytic sites, Glu200 and Glu298 with β-d-galactose (GAL) separately.
2 Materials and methods
2.1 Sequence retrieval
The amino acid sequence of β-galactosidase enzyme from A. niger was retrieved from GenPept database available at National Centre for Biotechnology Information (http://www.ncbi.nlm.nih.gov/protein/).
2.2 Comparative modeling and structural annotation
The prediction of protein structure using comparative modeling is an active and promising area of research. The comparative modeling of the target protein was performed using 3D jigsaw server (http://bmm.cancerresearchuk.org/~3djigsaw/) (Bates et al. 2001), validated using PROCHECK (Morris et al. 1992), and the structural annotation was described using SAS-sequence annotated by structure server (http://www.ebi.ac.uk/thornton-srv/databases/sas/) (Milburn et al. 1998), ProFunc-protein function from structure server (http://www.ebi.ac.uk/thornton-srv/databases/ProFunc/) (Laskowski et al. 2005), and PDBsum-pictorial database of 3D structures in the Protein Data Bank server (http://www.ebi.ac.uk/pdbsum/) (Laskowski 2001) available at European Bioinformatics Institute. Domain identification in the amino acid sequence of β-galactosidase enzyme from A. niger was performed using InterProScan tool (Bateman et al. 2004).
2.3 Structural comparison
Protein structure comparisons are often used to highlight the similarities and differences among related homologous-3D structures. Homologous proteins are descended from a common ancestral protein, but have subsequently duplicated, evolved along separate paths, and thus changed over time (Mark and Jukka 2000; Dwivedi et al. 2013). Structural comparison of the modeled structure was performed using Dali server (http://ekhidna.biocenter.helsinki.fi/dali_server/start) (Holm and Rosenström 2010) in PDB database to find out the similar structures in another organisms.
2.4 Molecular docking
The catalytic residue and their ligand interaction for the modeled structure identified using SAS server was used for molecular docking to find out the protein–ligand interaction using ArgusLab 4.0.1program (Thompson 2004).
3 Results
3.1 Sequence retrieval
The amino acid sequence of β-galactosidase enzyme from A. niger was retrieved in FASTA format using their accession number, AAC60538.1
3.2 Comparative modeling and structural annotation
The 3D structure of target protein was generated by 3D jigsaw server using comparative modeling approach. The predicted structure was visualized under PyMol program. (Fig. 1) The PROCHECK analysis of the modeled structure yielded the corresponding Ramachandran plot for the predicted structure (Fig. 2), showing the following anomalies: the ϕ/ψ angles of 89.1 % residues are in the most favoured regions, 10 % in the additional allowed regions, 0.6 % in the generously allowed regions, 0.2 % in the disallowed regions. Based on an analysis of 118 structures of resolution of at least 2.0 Å and R-factor no greater than 20 %, a good quality model would be expected to have over 90 % in the most favored regions. The wiring diagram was generated by SAS server as depicted in Fig. 3. The ProMotif documentation of the enzyme via Profunc server showed the results for the secondary structure summary as: the 966 residue span of the structure consisting of 226 residues (23.4 %), which were involved in the formation of the strands, 104 residues (10.8 %) for the alpha helices, 26 residues (2.7 %) for the 3–10 helix and 610 residues (63.1 %) for the several other structural moieties. Also, the result showed 15 β-sheets, 5 β-α-β motifs, 14 β-hairpins, 12 β-bulges, 52 strands, 20 helices, 12 helix–helix interactions, 109 β-turns, 9 γ-turns. Five distinct domains were found to compose the structural framework of the target protein. The first domain belonging to glycosyl hydrolases family 35 was found from Ser52 to Phe387 residue regions containing the catalytic site Glu200 (Rojas et al. 2004) and Glu298 (Maksimainen et al. 2011). The second domain belonging to β-galactosidase, domain 2 family was found from Tyr395 to Tyr573 residue regions. The third domain belonging to β-galactosidase, domain 3 family was found from Trp574 to Ala658 positions. The fourth and fifth β-galactosidase jelly roll domains belonging to β-galactosidase domain 4 and 5 families were found from Tyr690 to Leu802 residue regions. The topology of the enzyme structure is illustrated in Fig. 4.

3D structure of β-galactosidase enzyme in Aspergillus niger from PyMol

Ramachandran plot of β-galactosidase enzyme from Aspergillus niger derived from PROCHECK

The figure shows the wiring diagram of the β-galactosidase enzyme from Aspergillus niger, created via SAS server

Topology of the enzyme β-galactosidase showing the pattern alignment of the five constituent domains
3.3 Structural comparison
The structural comparison of the modeled structure showed highest similarity with crystal structure of Penicillium sp. β-galactosidase in PDB database.
3.4 Molecular docking
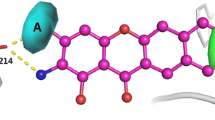
The catalytic residues (Glu200 and Glu298) and the interacting ligand (GAL) were identified using SAS server. The molecular docking of both the catalytic residues executed separately with GAL showed the best docking score in both the cases (Figs. 5, 6, 7, 8). The bond interactions between the catalytic sites and the ligand are shown in Table 1. Though the bond interactions are efficient at both the sites, the bond formed between the catalytic site Glu200 was found stronger due to two H-bonds in comparison to the other catalytic site Glu298.

The ball and stick representation of the molecular interaction between β-galactosidase active site Glu200 (dark green) and the ligand β-d-galactose (yellow) (color figure online)

The binding of β-d-galactose (red solid sphere) at the enzyme active site Glu200 of β-galactosidase (gray solid surface) (color figure online)

The figure shows the Ball and stick representation of the molecular docking between β-galactosidase active site Glu298 (bicolor) and the ligand beta-d-galactose (green) (color figure online)

The figure shows the binding of the ligand (red solid sphere) at the enzyme active site of β-galactosidase (grey solid surface) (color figure online)
4 Discussion
The modeled structure of β-galactosidase from A. niger resembled the high-resolution experimental structure of the same enzyme from Penicillium sp. Hence it could be evidently concluded that the range of functionality of the enzyme from A. niger could be enhanced. Also, β-d-galactose was found to behave as a ligand for the enzyme in A. niger. β-d-galactose was reported with strong bonding affinities for two different catalytic sites Glu200 and Glu298 of the enzyme, forming a greater association with Glu200 catalytic site. The molecular docking of β-d-galactose with β-galactosidase from A. niger was carried out to throw light into the stereotypic behavior of the ligand toward the enzyme binding sites. It was found that β-d-galactose behaved as a potent inhibitor of the enzyme. In cellular pool, where β-galactosidase is not available in its native form, this conclusion was quite far-fetched. Analytically, if β-d-galactose can be made to accumulate in the enzyme activity area, it would compete with the substrate and irreversibly bind with the active site of the enzyme and rendering it inactive for functioning, thereby, causing a feedback inhibition.
References
Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Eddy SR (2004) The Pfam protein families database. Nucleic Acids Res 32:D138–D141
Bates PA, Kelley LA, MacCallum RM, Sternberg MJE (2001) Enhancement of protein modelling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins 45(S5):39–46
Dimitris G,Hatzinikolaou, EfstathiosKatsifas, Diomi Mamma, Amalia D. Karagouni, Paul Christakopoulos, DimitrisKekos (2005) Modeling of the simultaneous hydrolysis–ultrafiltration of whey permeate by a thermostable β-galactosidase from Aspergillus niger. Biochem Eng J. 24(2):161–172
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92(20):9363–9367
Dorland NW (1997) Dorland’s illustrated medical dictionary, 28th edn. Saunders, Philadelphia
Dwivedi VD, Arora S, Pandey A (2013) Computational analysis of physico-chemical properties and homology modeling of carbonic anhydrase from Cordyceps militaris. Netw Model Anal Health Inform Bioinforma:1–4. doi:10.1007/s13721-013-0036-8
Holm L, Rosenström P (2010) Dali server: conservation mapping in 3D. Nucl Acids Res 38:W545–W549
Mark S. Johnson and Jukka V. Lehtonen (2000). Comparison of protein three-dimensional structures. Bioinformatics: sequence, structure, and databanks. Oxford University Press, p 15
Klich MA (2002) Identification of common Aspergillus species. Centraalbureau voor Schimmelcultures, Utrecht. ISBN 90-70351-46-3
Laskowski RA (2001) PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res 29(1):221–222
Laskowski RA, Watson JD, Thornton JM (2005) ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res 33:89–93
Maksimainen M, Hakulinen N, Kallio JM, Timoharju T, Turunen O, Rouvinen J (2011) Crystal structures of Trichoderma reesei β-Galactosidase reveal conformational changes in the active site. J Struct Biol 174:156–163
Milburn D, Laskowski RA, Thornton JM (1998) Sequences annotated by structure: a tool to facilitate the use of structural information in sequence analysis. Protein Eng 11(10):855–859
Mlichová Z, Rosenberg M (2006) Current trends of β-galactosidase application in food technology. J Food Nutr Res 2(45):47–54
Morris AL, MacArthur MW, Hutchinson EG, Thornton JM (1992) Stereochemical quality of protein structure coordinates. Proteins 12:345–364
Rojas AL, Nagem RAP, Neustroev KN, Arand M, Adamska M, Eneyskaya EV, Kulminskaya AA, Garratt RC, Golubev AM, Polikarpov I (2004) Crystal structures of β-galactosidase from Penicillium sp. and its complex with galactose. J Mol Biol 343(5):1281–1292
Staiano M, Bazzicalupo P, Rossi M, D’Auria S (2005) Glucose biosensors as models for the development of advanced protein-based biosensors. Mol BioSyst 354(1). doi:10.1039/B513385H
Thompson MA (2004) Molecular docking using ArgusLab, an efficient shape-based search algorithm and the AScore scoring function. ACS meeting, Philadelphia, 172(42)
Acknowledgments
We are grateful to Dr. Sarad Kumar Mishra, Department of Biotechnology, D.D.U. Gorakhpur University, Gorakhpur, India for his kind support and necessary suggestions whenever needed.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ghosh, S.K., Pandey, A., Arora, S. et al. Comparative modeling and docking studies of β-galactosidase from Aspergillus niger . Netw Model Anal Health Inform Bioinforma 2, 297–302 (2013). https://doi.org/10.1007/s13721-013-0046-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13721-013-0046-6