
Abstract
Global vaccination effort and better understanding of treatment strategies provided a ray of hope for improvement in COVID-19 pandemic, however, in many countries, the disease continues to collect its death toll. The major pathogenic mechanism behind severe cases associated with high mortality is the burst of pro-inflammatory cytokines TNF, IL-6, IFNγ and others, resulting in multiple organ failure. Although the exact contribution of each cytokine is not clear, we provide an evidence that the central mediator of cytokine storm and its devastating consequences may be TNF. This cytokine is known to be involved in activated blood clotting, lung damage, insulin resistance, heart failure, and other conditions. A number of currently available pharmaceutical agents such as monoclonal antibodies and soluble TNF receptors can effectively prevent TNF from binding to its receptor(s). Other drugs are known to block NFkB, the major signal transducer molecule used in TNF signaling, or to block kinases involved in downstream activation cascades. Some of these medicines have already been selected for clinical trials, but more work is needed. A simple, rapid, and inexpensive method of directly monitoring TNF levels may be a valuable tool for a timely selection of COVID-19 patients for anti-TNF therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
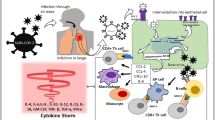
Respiratory distress and activation of blood clotting in severe COVID-19 cases result in unusually high mortality rates, particularly among people of advanced age and those that have comorbidities—cardiovascular or pulmonary disease, obesity, and diabetes. Severe disease is associated with “cytokine storm”, a delayed onset burst of pro-inflammatory cytokines in circulation. The cytokines associated with fatalities are TNF, IL-6, IL-8, IFNγ and possibly others [1]. It is difficult to identify the pivotal cytokine(s) in this process, but some facts argue in favor of TNF.
TNF is involved in pathogenesis of comorbidities linked to severe COVID-19 disease
Numerous pathologies are associated with elevated TNF levels, from autoimmune disorders to sepsis and cancer. In the respiratory system, TNF causes bronchial hyperreactivity, narrowing of the airways, damage to the respiratory epithelium, stimulation of collagen synthesis and fibrosis [2, 3]. Chronic obstructive pulmonary disease (COPD) is a known risk factor for severe COVID-19 disease [4]. Circulating TNF levels are increased in COPD [5]. The role of TNF in this disease has been suggested, and TNF inhibition was shown effective in lowering the incidence of hospitalization in one study [6] but did not improve health status and lung function in the other [7]. However, TNF blockage in COVID-19 patients with COPD may be advocated as a measure to reduce additive damage to already compromised lungs. In addition, pulmonary fibrosis is observed in a significant proportion of patients after acute COVID-19 pneumonia [8]. Although the role of TNF in this process is not established, there is evidence for TNF involvement in a closely related idiopathic pulmonary fibrosis [9]. Administration of anti-TNF drugs during the acute phase of infection may subsequently alleviate development of this complication.
The effects of TNF on the cardiovascular system are also well known. TNF significantly contributes to the development of heart failure by direct negative inotropic and pro-apoptotic effects on cardiomyocytes, and by other mechanisms [10]. TNF is also elevated in patients with hypertension [11]. Moreover, TNF levels are increased in obesity, and TNF is considered to play a role in insulin resistance [12, 13]. All these conditions are risk factors for development of severe COVID-19 disease and associated mortality or long-term complications.
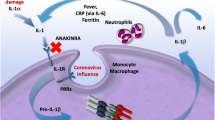
The ability of TNF to activate tissue factor on endothelial cells and monocytes and induce severe blood clotting during infection has been well documented [14,15,16,17,18]. TNF also inhibits fibrinolysis by increasing plasminogen activator inhibitor [19]. Reports on pro-coagulant activities induced by IL-6 are scarce [20, 21]. Increased blood clotting observed in COVID-19 patients is a well- documented complication requiring anti-coagulant therapy.
Both TNF and IL-6 levels are elevated with age: this chronic inflammation termed inflammaging is suggested to serve as a biomarker of frailty and mortality in elderly population [22]. Age-related loss of muscle mass and strength is particularly attributed to the action of TNF [23], and exposure of human cells to TNF in vitro can induce cell senescence [24]. Strong association of TNF with ageing may explain, to some extent, higher incidence of severe COVID-19 disease in patients of advanced age. Interestingly, mTOR inhibitor has been suggested recently for treatment of severe disease based on its ability to alleviate cytokine storm [25]. The drug is also known to improve longevity and reverse age-related immunosenescence in experimental animals, and its use in older adults may prevent age-associated complications of COVID-19 by poorly understood “rejuvenating” mechanisms [26]. On the other hand, the effects of mTOR inhibition may be reduced to a direct inhibition of TNF synthesis or signal transduction [27, 28].
Pro-inflammatory cytokines TNF, IL-6 and others are elevated in major depressive disorder, which is strongly associated with COVID-19 infection [29, 30]. TNF blockade using adalimumab in patients with rheumatoid arthritis with depression reduced serotonin transporter in the brain and improved the depression score [31]. On the other hand, a number of reports demonstrate anti-inflammatory effect of various antidepressants [30, 32]. Of interest is a retrospective multicenter study reported by a French group demonstrating that antidepressants reduce the risk of intubation and death in hospitalized patients with COVID-19 [33]. At least two clinical trials are currently underway to investigate the impact of this class of drugs on the disease outcomes (NCT04342663, NCT04377308).
Both TNF and IL-6 levels may serve as independent predictors of COVID-19 severity. However, only TNF but not IL-6 levels were higher in COVID patients with diabetes, hypertension, chronic kidney disease and chronic heart failure, thus providing a strong argument of TNF being a central factor for severe disease in these individuals [34]. In addition, in contrast to TNF, systemically administered IL-6 is fairly well tolerated [35], again suggesting that elevation of TNF rather than IL-6 is responsible for the pathology observed in complicated COVID-19 scenarios. Although some studies suggest that anti-IL-6 monoclonal antibody Tocilizumab is beneficial for the patients with severe COVID-19, other reports did not support that observation [36]. Similarly, clinical trials with Sarilumab, an IL-6 receptor blocker, did not meet primary and secondary endpoints in hospitalized COVID-19 patients [37]. Future clinical studies are believed to shed more light on particular roles of these two cytokines in the disease.
Factors that activate TNF
Various factors activate production of TNF. Signals from toll-like receptors (TLRs), T cell and B cell receptors to antigens (TCR, BCR), receptors to Fc fragments of immunoglobulins (FcRs), cytokines (IL-1, IL-2, IFNγ, GM-CSF, TNF itself), mitogens, superantigens, radiation, osmotic stress, high glucose levels, microbes and their products induce TNF [38]. TNF production in response to a “danger signal” is very rapid. Activation of innate immunity through pattern recognition receptors, such as TLRs, results in a rapid release of TNF which is followed by IL-6, and blocking TNF attenuates IL-6 levels, again, suggesting that IL-6 is a secondary cytokine in the cytokine cascade [39]. Single-stranded RNA of SARS-CoV2 should be recognized by TLR7, TLR8, and TLR3. It is of a particular interest that SARS-CoV2 has RNA genome of 30,000 bp, the largest among RNA viruses (for comparison, Influenza A virus has a genome of only 13,500 bp) [40]. One can speculate that the size of RNA may contribute to an enhanced innate immune response to the virus resulting in the cytokine storm.
Blocking TNF upstream of TNF receptors
TNF blocking agents have been used for two decades to treat various inflammatory conditions and have a good safety record. The most well studied are monoclonal anti-TNF neutralizing antibodies (mNAbs) and soluble TNF Receptor (TNFR)-Fc fusion protein. Both drugs have FDA approval for treatment of several autoimmune diseases, and both drugs prevent interaction of TNF with its receptors TNFR1 and TNFR2. Anti-TNF drugs have been suggested for the treatment of SARS in 2004 [41], and accumulating evidence indicates that TNF blockade is beneficial for COVID-19 patients. First, a small study from Germany demonstrated a significant therapeutic effect of anti-TNF mNAb on the outcomes in severe COVID-19 cases in hospitalized patients [42]: mortality rate was 14% in anti-TNF-treated group compared to 35% in control group. Then, preliminary data of 600 cases from 40 countries have been published and indicated that anti-TNF monotherapy for rheumatic diseases was associated with a reduced risk of hospitalization of COVID-19 patients (odds ratio = 0.40) [43]. Later, a meta-analysis of more than 6000 cases revealed that TNF inhibitors administered to patients with immune-mediated inflammatory diseases alone were better protectors from severe COVID-19 associated hospitalizations and deaths than azathioprine/6-mercaptopurine or methotrexate [44]. Another study reported that TNF blockers administered to patients with rheumatoid arthritis and spondyloarthropathies significantly (by > 80%) reduced the risk of COVID-19 infection [45].
Moreover, based on the data from SECURE-IBD database a beneficial effect of anti-TNF mNAbs was reported in COVID-19 patients with inflammatory bowel disease: namely, only 30 out of 198 (15%) patients with IBD receiving anti-TNF therapy required hospitalization for COVID-19, and only 3% of them died, whereas 67% IBD patients on steroid therapy were hospitalized and 25% of them died [44]. Finally, Kridin et al. recently analyzed outcomes of COVID-19 in patients with psoriasis treated with anti-TNF biologics and found that TNF blockade significantly decreased the risk of COVID-19 associated hospitalization. The authors did not see reduction in mortality, but mortality events were very low in this study [45]. These observations also suggest that immunosuppressive properties of TNF blockers do not exacerbate viral disease due to a weakened clearance of the pathogen.
Blocking TNF signals downstream of TNFRs
The major signal transduction pathway activated by TNF binding to its receptors involves nuclear factor kappa B (NFkB). Therefore, it is reasonable to suggest that many effects of TNF may be blocked at the level downstream of the receptors by NFkB inhibitors. In addition to mTOR inhibitor mentioned earlier, numerous known and potential drugs have been found to inhibit NFkB [46, 47]. Aspirin and sodium salicylate inhibit activation of NFkB by blocking IkB kinase [48]. Glucocorticoids suppress expression of inflammatory genes by binding glucocorticoid receptor with NFkB, and increasing expression of inhibitory protein of NFkB, IkBa. Sulfasalazine and gold compounds also inhibit NFkB activation and were suggested for treatment of rheumatoid arthritis [49].
Well known cardiac glycosides inhibit NFkB which in turn decreases production of inflammatory cytokines [50]. These drugs also demonstrate anti-viral activity to SARS-CoV-2 by inhibiting membrane Na–K ATPase, used by RNA viruses for entering the cell, and by inhibiting translation, which makes them very attractive in COVID-19 patients with cardiac insufficiency [51,52,53].
Nifedipine, a blocker of calcium channels used as an antihypertensive also inhibits NFkB. In an experimental rat model of vascular lung leakage induced by high altitude hypoxia, a condition that has much in common with pulmonary damage in COVID-19 patients, the drug inhibited vascular leakage, oxidative stress, and reduced inflammatory cytokine production [54].
Another class of antihypertensive drugs, ACE inhibitors, also suppress NFkB and inflammation, reduce blood pressure, and limit viral entry [55, 56]. This is particularly interesting since ACE2 is the major receptor for SARS-CoV-2 Spike glycoprotein, and several studies demonstrated that the use of ACE inhibitors was associated with a reduced risk of severe COVID‐19 disease, morbidity, and mortality probably due to a direct inhibition of viral Spike protein binding to ACE2 [57, 58].
Bortezomib is a proteasome and NFkB inhibitor used for treatment of multiple myeloma [59]. It has been recently suggested that this class of drugs may also be used as a therapy for COVID-19 because it interferes with SARS-CoV-2 entry into eucaryotic cells, inhibits RNA and protein synthesis and viral replication, induces endoplasmic reticulum stress, and alleviates cytokine production [60].
A large group of kinase inhibitors initially introduced for treatment of malignancies should also be considered. Tyrosine kinase inhibitor imatinib, for instance, has been shown to down regulate several inflammatory cytokines and NFkB levels [61, 62]. A successful case report on administration of imatinib for treatment of a progressing COVID-19 pneumonia has been published recently: 3 days after administration of the drug at 400 mg once daily, the patient’s body temperature normalized, and oxygen treatment was discontinued. The patient was discharged on day 16 and remained asymptomatic with a significant improvement on chest radiograph on day 20 after discharge [63].
Janus kinases (JAKs) participate in signal transduction following cytokine binding to their receptors, including pro-inflammatory cytokines [64]. JAK inhibitors have been shown to reduce serum IL-6, IFNγ, IL-1, IL-17 and TNF levels in a way similar to TNF blockers in patients with rheumatoid arthritis and ulcerative colitis [65,66,67]. In vitro, they inhibit TNF-induced NFkB activation in human macrophages [68]. JAK inhibitors have also been suggested as a novel therapy for COVID-19 and have been used by several groups based on their ability to reduce both viral entry and cytokine production [69]. Beneficial effects of JAK inhibitors in human disease have been reported, including reduction of TNF and other cytokine levels [70, 71]. Baricitinib in combination with antiviral Remdesivir has been issued an emergency authorization by FDA as a treatment for hospitalized patients [72,73,74]. Noteworthy, another JAK inhibitor added to standard therapy has been reported effective in hospitalized COVID-19 patients with pneumonia in a multicenter randomized, placebo-controlled trial: the cumulative incidence of death or respiratory failure through day 28 was 18.1% in the JAK inhibitor group and 29.0% in the placebo group (risk ratio, 0.63; 95% confidence interval [CI], 0.41–0.97; P = 0.04) [75].
Who should receive anti-TNF therapy, and when?
The most optimal biomarker for use as a criterion to start anti-TNF/anti-NFkB/anti-kinase therapy is yet to be established. Direct measurement and subsequent monitoring of circulating TNF levels using express tests, such as currently unavailable immunochromatography, seems to be the method of choice, since it can be used in an outpatient setting. However, other biomarkers such as IL-6 may be used as long as they reflect the brewing of cytokine storm. The decision to initiate anti-TNF therapy should be made in COVID-19 patients admitted to the hospital [76] and considering each patient’s presentation of symptoms and complications. Clinical trials on TNF blockade have started in Oxford and Tufts Universities, but more studies could significantly advance our understanding of which of a variety of available drugs are most likely to improve outcomes [76,77,78]. Although TNF/NFkB inhibitors are considered relatively safe even when administered in a long term, a safety concern should always be kept in mind considering TNF inhibitors are immunosuppressive.
Concluding remarks
-
TNF is involved in pathogenesis of severe COVID-19 and may play a central role in “cytokine storm”.
-
TNF or its downstream mediators may be inhibited by administration of existing drugs: direct TNF binders, NFkB blockers, and kinase inhibitors.
-
Clinical trials are urgently needed to test different TNF/NFkB/kinase inhibitors for treatment or prevention of severe COVID-19 cases, and to establish indications and end-point criteria for such treatment.
-
Rapid tests like lateral flow/immunochromatography for measurement of TNF or other biomarkers may be invaluable tools for timely assessment of patients and decision making in administration of TNF inhibitors.
-
Although this paper may seem biased towards the benefits of anti-TNF therapy, the bias is intentional, as the review aims to draw attention of the reader to a possible role of TNF in COVID-19. The limited space did not allow us to extensively discuss other aspects of COVID-19 pathogenesis.
-
In heterogeneous human population, TNF may be up-regulated to a different degree, and its detrimental effects may be different due to different sensitivity of each individual, comorbidities, and other factors. Currently, it is difficult to establish any threshold after which an individual patient will inevitably deteriorate. Probably, the only way of learning that is a clinical trial, as mentioned earlier.
Data availability
All data for this manuscript is available in public electronic databases.
References
Costela-Ruiz VJ, Illescas-Montes R, Puerta-Puerta JM, Ruiz C, Melguizo-Rodríguez L (2020) SARS-CoV-2 infection: the role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev 54:62–75. https://doi.org/10.1016/j.cytogfr.2020.06.001 (Epub 2020 Jun 2)
Mukhopadhyay S, Hoidal JR, Mukherjee TK (2006) Role of TNFalpha in pulmonary pathophysiology. Respir Res 7:125. https://doi.org/10.1186/1465-9921-7-125
Malaviya R, Laskin JD, Laskin DL (2017) Anti-TNFα therapy in inflammatory lung diseases. Pharmacol Ther 180:90–98. https://doi.org/10.1016/j.pharmthera.2017.06.008 (Epub 2017 Jun 19)
Leung JM, Niikura M, Yang CWT, Sin DD (2020) COVID-19 and COPD. Eur Respir J 56:2002108. https://doi.org/10.1183/13993003.02108-2020
Yao Y, Zhou J, Diao X, Wang S (2019) Association between tumor necrosis factor-α and chronic obstructive pulmonary disease: a systematic review and meta-analysis. Ther Adv Respir Dis. https://doi.org/10.1177/1753466619866096
Suissa S, Ernst P, Hudson M (2008) TNF-alpha antagonists and the prevention of hospitalisation for chronic obstructive pulmonary disease. Pharmacol Ther 21:234–238. https://doi.org/10.1016/j.pupt.2007.03.003 (Epub 2007 Apr 11)
Rennard SI, Fogarty C, Kelsen S, Long W, Ramsdell J, Allison J, Mahler D, Saadeh C, Siler T, Snell P, Korenblat P, Smith W, Kaye M, Mandel M, Andrews C, Prabhu R, Donohue JF, Watt R, Lo KH, Schlenker-Herceg R, Barnathan ES, Murray J (2007) The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 175:926–934
George PM, Wells AU, Jenkins RG (2020) Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir Med 8:807–815
Lechowicz K, Drozdzal S, Machaj F, Rosik J, Szostak B, Zegan-Baranska M (2020) COVID-19: the potential treatment of pulmonary fibrosis associated with SARS-CoV-2 infection. J Clin Med. https://doi.org/10.3390/jcm9061917
Hanna A, Frangogiannis NG (2020) Inflammatory cytokines and chemokines as therapeutic targets in heart failure. Cardiovasc Drugs Ther 34:849–863. https://doi.org/10.1007/s10557-020-07071-0 (Epub 2020 Sep 9)
De Miguel C, Rudemiller NP, Abais JM, Mattson DL (2015) Inflammation and hypertension: new understandings and potential therapeutic targets. Curr Hypertens Rep 17:507. https://doi.org/10.1007/s11906-014-0507-z
Kanneganti TD, Dixit VD (2012) Immunological complications of obesity. Nat Immunol 13:707–712
Harder-Lauridsen NM, Krogh-Madsen R, Holst JJ, Plomgaard P, Leick L, Pedersen BK, Fischer CP (2014) Effect of IL-6 on the insulin sensitivity in patients with type 2 diabetes. Am J Physiol Endocrinol Metab 306:E769–E778. https://doi.org/10.1152/ajpendo.00571.2013 (Epub 2014 Jan 28)
Nawroth PP, Stern DM (1986) Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J Exp Med 163:740–745
Conckling PR, Greenberg CS, Weinberg JB (1988) Tumor necrosis factor induces tissue factor-like activity in human leukemia cell line U937 and peripheral blood monocytes. Blood 72:128–133
Schwager I, Jungi TW (1994) Effect of human recombinant cytokines on the induction of macrophage procoagulant activity. Blood 83:152–160
Page EM, Ariëns RAS (2021) Mechanisms of thrombosis and cardiovascular complications in COVID-19. Thromb Res 200:1–8. https://doi.org/10.1016/j.thromres.2021.01.005
Bautista-Vargas M, Bonilla-Abadía F, Cañas CA (2020) Potential role for tissue factor in the pathogenesis of hypercoagulability associated with in COVID-19. J Thromb Thrombolysis 50:479–483. https://doi.org/10.1007/s11239-020-02172-x
Schleef RR, Bevilaqua MP, Sawdey M, Gimbrone MA, Loskutoff DJ (1988) Cytokine activation of vascular endothelium. Effects on tissue-type plasminogen activator and type 1 plasminogen activator inhibitor. J Biol Chem 263:5797–5803
Neumann FJ, Ott I, Marx N, Luther T, Kenngott S, Gawaz M et al (1997) Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arterioscler Thromb Vasc Biol 17:3399–3405
Kang S, Tanaka T, Inoue H, Ono C, Hashimoto S, Kioi Y et al (2020) IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci U S A 117:22351–22356. https://doi.org/10.1073/pnas.2010229117
Michaud M, Balardy L, Moulis G, Gaudin C, Peyrot C, Vellas B, Cesari M, Nourhashemi F (2013) Proinflammatory cytokines, aging, and age-related diseases. J Am Med Dir Assoc 14:877–882
Reid MB, Li YP (2001) Tumor necrosis factor-alpha and muscle wasting: a cellular perspective. Respir Res 2:269–272. https://doi.org/10.1186/rr67 (Epub 2001 Jul 12)
Mavrogonatou E, Konstantinou A, Kletsas D (2018) Long-term exposure to TNF-alpha leads human skin fibroblasts to a p38 MAPK- and ROS-mediated premature senescence. Biogerontology 19(3–4):237–249. https://doi.org/10.1007/s10522-018-9753-9 (Epub 2018 Mar 26)
Omarjee L, Janin A, Perrot F, Laviolle B, Meilhac O, Mahe G (2020) Targeting T-cell senescence and cytokine storm with rapamycin to prevent severe progression in COVID-19. Clin Immunol 216:108464. https://doi.org/10.1016/j.clim.2020.108464
Bischof E, Siow RC, Zhavoronkov A, Kaeberlein M (2021) The potential of rapalogs to enhance resilience against SARS-CoV-2 infection and reduce the severity of COVID-19. Lancet Healthy Longev 2(2):e105–e111. https://doi.org/10.1016/S2666-7568(20)30068-4
Giordano A, Avellino R, Ferraro P, Romano S, Corcione N, Romano MF (2006) Rapamycin antagonizes NF-kappaB nuclear translocation activated by TNF-alpha in primary vascular smooth muscle cells and enhances apoptosis. Am J Physiol Heart Circ Physiol 290:H2459–H2465. https://doi.org/10.1152/ajpheart.00750.2005 (Epub 2006 Jan 20)
Park JW, Jeon YJ, Lee JC, Ahn SR, Ha SW, Bang SY et al (2012) Destabilization of TNF-alpha mRNA by Rapamycin. Biomol Ther (Seoul) 20:43–49. https://doi.org/10.4062/biomolther.2012.20.1.043
Cothran TP, Kellman S, Singh S, Beck JS, Powell KJ, Bolton CJ et al (2020) A brewing storm: The neuropsychological sequelae of hyperinflammation due to COVID-19. Brain Behav Immun 88:957–958
Kopschina Feltes P, Doorduin J, Klein HC, Juárez-Orozco LE, Dierckx RAJO, Moriguchi-Jeckel CM, de Vries EFJ (2017) Anti-inflammatory treatment for major depressive disorder: implications for patients with an elevated immune profile and non-responders to standard antidepressant therapy. J Psychopharmacol 31:1149–1165. https://doi.org/10.1177/0269881117711708
Cavanagh J, Paterson C, McLean J, Pimlott S, McDonald M, Patterson J et al (2010) Tumour necrosis factor blockade mediates altered serotonin transporter availability in rheumatoid arthritis: a clinical, proof-of-concept study. Ann Rheum Dis 69:1251–1252
Pashaei Y (2021) Drug repurposing of selective serotonin reuptake inhibitors: Could these drugs help fight COVID-19 and save lives? J Clin Neurosci 88:163–172
Hoertel N, Rico MS, Vernet R, Beeker N, Jannot A-S, Neuraz A et al (2021) Association between SSRI antidepressant use and reduced risk of intubation or death in hospitalized patients with COVID- 2019: results from an observational study. Mol Psychiatry 26:5199–5212
Del Valle DM, Kim-Schulze S, Huang H-H, Beckmann ND, Nirenberg S, Wang B et al (2020) An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med 26:1636–1643
Stouthard JML, Romijn JA, van der Poll T, Endert E, Klein S, Bakker PJM et al (1995) Endocrine and metabolic effects of interleukin-6 in humans. Am J Physiol 268:E813–E819
Hamidreza Samaee H, Mohsenzadegan M, Ala S, Maroufi SS, Moradimajd P (2020) Tocilizumab for treatment patients with COVID-19: Recommended medication for novel disease. Int Immunopharmacol 89:107018
Sanofi press release (2020). https://www.sanofi.com/en/media-room/press-releases/2020/2020-09-01-07-00-00. Accessed 14 Jan 2022
Falvo JV, Tsytsykova AV, Goldfeld AE (2010) Transcriptional Control of the TNF Gene. Curr Dir Autoimmun 11:27–60
Ghezzi P, Sacco S, Agnello D, Marullo A, Caselli G, Bertini R (2000) Lps induces IL-6 in the brain and in serum largely through TNF production. Cytokine 12:1205–1210. https://doi.org/10.1006/cyto.2000.0697
Fehr AR, Perlman S (2015) Coronaviruses: an overview of their replication and pathogenesis. Coronaviruses 1282:1–23. https://doi.org/10.1007/978-1-4939-2438-7_1
Tobinick E (2004) TNF-α inhibition for potential therapeutic modulation of SARS coronavirus infection. Curr Med Res Opin 20:39–40. https://doi.org/10.1185/030079903125002757
Stallmach A, Kortgen A, Gonnert F, Coldewey SM, Reuken P, Bauer M (2020) Infliximab against severe COVID-19-induced cytokine storm syndrome with organ failure-a cautionary case series. Crit Care 24:444. https://doi.org/10.1186/s13054-020-03158-0
Gianfrancesco M, Hyrich KL, Al-Adely S, Carmona L, Danila MI, Gossec L et al (2020) Characteristics associated with hospitalisation for COVID-19 in people with rheumatic disease: data from the COVID-19 global rheumatology alliance physician-reported registry. COVID-19 Global rheumatology alliance. Ann Rheum 79:859–866. https://doi.org/10.1136/annrheumdis-2020-217871
Izadi Z, Brenner EJ, Mahil SK, Dand N, Yiu ZZN, Yates M et al (2021) Association between tumor necrosis factor inhibitors and the risk of hospitalization or death among patients with immune-mediated inflammatory disease and COVID-19. JAMA Netw Open 4:e2129639. https://doi.org/10.1001/jamanetworkopen.2021.29639
Salesi M, Shojaie B, Farajzadegan Z, Salesi N, Mohammadi E (2021) TNF-α blockers showed prophylactic effects in preventing COVID-19 in patients with rheumatoid arthritis and seronegative spondyloarthropathies: a case-control study. Rheumatol Ther 8:1355–1370
Tursi A, Vetrone LM, Papa A (2020) Anti-TNF-α agents in inflammatory bowel disease and course of COVID-19. Inflamm Bowel Dis 26:e73. https://doi.org/10.1093/ibd/izaa114
Kridin K, Schonmann Y, Tzur Bitan D, Damiani G, Peretz A, Weinstein O, Cohen AD (2021) Coronavirus disease COVID-19-associated hospitalization and mortality in patients with psoriasis: a population-based study. Am J Clin Dermatol 22:709–718. https://doi.org/10.1007/s40257-021-00605-8
Gilmore TD, Herscovitch M (2006) Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 25:6887–6899
Miller SC, Huang R, Sakamuru S, Shukla SJ, Attene-Ramos MS, Shinn P et al (2010) Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem Pharmacol 79:1272–1280
Kopp E, Ghosh S (1994) Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 265:956–959
Jue DM, Jeon KI, Jeong JY (1999) Nuclear factor kappaB (NF-kappaB) pathway as a therapeutic target in rheumatoid arthritis. J Korean Med Sci 14:231–238
Yang Q, Huang W, Jozwik C, Lin Y, Glasman M, Caohuy, et al (2005) Cardiac glycosides inhibit TNF-alpha/NF-kappaB signaling by blocking recruitment of TNF receptor-associated death domain to the TNF receptor. Proc Natl Acad Sci U S A 102:9631–9636
Siniorakis E, Arvanitakis S, Elkouris M (2020) Cardiac glycosides and COVID-19: would it be a promising therapeutic approach? An Acad Bras Cienc 92:e20201080. https://doi.org/10.1590/0001-3765202020201080
Haux J (2020) Digitalis for coronavirus infection! BMJ 368:m1252
Amarelle L, Katzen J, Shigemura M, Welch LC, Cajigas H, Peterlanderi C et al (2019) Cardiac glycosides decrease influenza virus replication by inhibiting cell protein translational machinery. Am J Physiol Lung Cell Mol Physiol 316:L1094–L1106
Sarada SKS, Veeramohan HP, Titto M, Saumya S, Chitharanjan M (2012) Nifedipine inhibits hypoxia induced transvascular leakage through down regulation of NFkB. Respir Physiol Neurobiol 183:26–34. https://doi.org/10.1016/j.resp.2012.05.016 (Epub 2012 May 22)
Hernandez-Presa MA, Bustos C, Ortego M, Tuñón J, Ortega L, Egido J (1998) ACE inhibitor quinapril reduces the arterial expression of NF-kappaB-dependent proinflammatory factors but not of collagen I in a rabbit model of atherosclerosis. Am J Pathol 153:1825–1837
Hrenak J, Simko F (2020) Renin-angiotensin system: an important player in the Pathogenesis of acute respiratory distress syndrome. Int J Mol Sci 21(21):8038. https://doi.org/10.3390/ijms21218038
Zhang P, Zhu L, Cai J, Lei F, Qin JJ, Xie J et al (2020) Association of inpatient use of angiotensin converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circ Res 126:1671–1681. https://doi.org/10.1161/CIRCRESAHA.120.317134
Meng J, Xiao G, Zhang J, He X, Ou M, Bi J et al (2020) Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg Microbes Infect 9:757–760. https://doi.org/10.1080/22221751.2020.1746200
Pascal L, Gay J, Willekens C, Wemeau M, Balkaran S, Robu D et al (2009) Bortezomib and Waldenstrom’s macroglobulinemia. Expert Opin Pharmacother 10:909–916. https://doi.org/10.1517/14656560902800160
Longhitano L, Tibullo D, Giallongo C, Lazzarino G, Tartaglia N, Galimberti S et al (2020) Proteasome inhibitors as a possible therapy for SARS-CoV-2. Int J Mol Sci 21:3622. https://doi.org/10.3390/ijms21103622 (PMID: 32443911)
Ciarcia R, Vitiello MT, Galdiero M, Pacilio C, Iovane V, d’Angelo D et al (2012) Imatinib treatment inhibit IL-6, IL-8, NF-KB and AP-1 production and modulate intracellular calcium in CML patients. J Cell Physiol 227:2798–2803
Weisberg E, Parent A, Yang PL, PL, Sattler M, Liu Q, et al (2020) Repurposing of kinase inhibitors for treatment of COVID-19. Pharm Res 37:167. https://doi.org/10.1007/s11095-020-02851-7 (PMID: 32778962)
Morales-Ortega A, Bernal-Bello D, Llarena-Barroso C, Frutos-Pérez B, Duarte-Millán MÁ, García de Viedma-García V et al (2020) Imatinib for COVID-19: a case report. Clin Immunol 218:108518. https://doi.org/10.1016/j.clim.2020.108518 (Epub 2020 Jun 27)
Roskoski R Jr (2016) Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol Res 111:784–803
Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez JB, Dowty ME et al (2016) The mechanism of action of tofacitinib - an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol 34:318–328 (Epub 2016 Mar 10)
Hanauer S, Panaccione R, Danese S, Cheifez A, Reinisch W, Higgins PDR et al (2019) Tofacitinib induction therapy reduces symptoms within 3 days for patients with ulcerative colitis. Clin Gastroenterol Hepatol 17:139–147. https://doi.org/10.1016/j.cgh.2018.07.009 (Epub 2018 Sep 10)
Li Y, Yuan L, Yang J, Lei Y, Zhang H, Xia L et al (2019) Changes in serum cytokines may predict therapeutic efficacy of tofacitinib in rheumatoid arthritis. Mediators Inflamm 2019:5617431. https://doi.org/10.1155/2019/5617431.eCollection2019
Yarilina A, Xu K, Chan C, Ivashkiv LB (2012) Regulation of inflammatory responses in tumor necrosis factor-activated and rheumatoid arthritis synovial macrophages by JAK inhibitors. Arthritis Rheum 64:3856–3866. https://doi.org/10.1002/art.37691
Jorgensen SCJ, Tse CLY, Burry L, Dresser LD (2020) Baricitinib: a review of pharmacology, safety, and emerging clinical experience in COVID-19. Pharmacotherapy 40:843–856. https://doi.org/10.1002/phar.2438 (Epub 2020 Jul 27)
Cantini F, Niccoli L, Nannini C, Matarrese D, Natale MED, Lotti P et al (2020) Beneficial impact of Baricitinib in COVID-19 moderate pneumonia; multicentre study. J Infect 81:647–679. https://doi.org/10.1016/j.jinf.2020.06.052 (Epub 2020 Jun 24)
Bronte V, Ugel S, Tinazzi E, Vella A, De Sanctis F, Canè S et al (2020) Baricitinib restrains the immune dysregulation in patients with severe COVID-19. J Clin Invest 130:6409–6416. https://doi.org/10.1172/JCI141772
https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-drug-combination-treatment-covid-19?utm_medium=email&utm_source=govdelivery. Accessed 14 Jan 2022
Guimarães PO, Quirk D, Furtado RH, Maia LN, Saraiva JF, Antunes MO et al (2021) STOP-COVID Trial Investigators. Tofacitinib in Patients Hospitalized with Covid-19 Pneumonia. N Engl J Med 385:406–415. https://doi.org/10.1056/NEJMoa2101643
Feldmann M, Maini RN, Woody JN, Holgate ST, Winter G, Rowland M et al (2020) Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 395:1407–1409. https://doi.org/10.1016/S0140-6736(20)30858-8
Robinson PC, Richards D, Tanner HL, Feldmann M (2020) Accumulating evidence suggests anti-TNF therapy needs to be given trial priority in COVID-19 treatment. Lancet Rheumatol 2:e653–e655. https://doi.org/10.1016/S2665-9913(20)30309-X
Robinson PC, Liew DFL, Liew JW, Monaco C, Richards D, Shivakumar S et al (2020) The potential for repurposing anti-TNF as a therapy for the treatment of COVID-19. Med (NY) 1:90–102. https://doi.org/10.1016/j.medj.2020.11.005
Acknowledgements
The authors would like to thank John Kamerud, Will Somers, Cara Williams, Thomas Wynn, and Jeremy Gale for fruitful discussions. VA is expressing his special appreciation to Marina Tovbina for the idea and inspiration.
Disclaimer
The purpose of this publication is merely educational. It is based on a personal opinion of the Authors, and does not reflect the research, development, and marketing strategies of Pfizer Inc. This publication should not be considered a request for proposal, a call for grant applications, or an encouragement to an off-label use of any drugs.
Funding
This study was sponsored by Pfizer Inc.
Author information
Authors and Affiliations
Contributions
VA wrote the manuscript and is the author of concept, CL wrote and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors Vitaly Ablamunits and Christopher Lepsy are employed by Pfizer.
Ethical approval
The manuscript has been internally reviewed and approved at Pfizer.
Informed consent
Not applicable.
Consent for publication
Both authors gave their consent to publish the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ablamunits, V., Lepsy, C. Blocking TNF signaling may save lives in COVID-19 infection. Mol Biol Rep 49, 2303–2309 (2022). https://doi.org/10.1007/s11033-022-07166-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07166-x