
Summary
Insulin like growth factor receptor (IGF-1R) targeting became one of the most investigated areas in anticancer drug development during the last decade. Strategies aiming to block IGF-1R activity include monoclonal antibodies, tyrosine kinase inhibitors and anti-ligands antibodies. Initial enthusiasm quickly encountered challenges. Unfortunately the validation of the efficacy of IGF-1R targeted agents in large clinical trials failed, however anecdotal single agent activity was seen in early studies. Consequently, questions regarding the selection of right target population and the appropriate trial design are arising. Despite the plethora of clinical trials conducted no predictive biomarker has been validated so far and resistance mechanisms to IGF-1R inhibitors remain unclear. The other issue to be addressed is how to best combine IGF-1R inhibitors with other therapeutic approaches. This review highlights the most relevant clinical data emphasizing the main tumor types where IGF-1R inhibition showed potential interest. We also tried to extract based on clinical and translational data some candidate biomarkers that could help better to select patient population who potentially could benefit most from this therapeutic approach.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Insulin like growth factor receptor—1 (IGF-1R) is a transmembrane receptor with tyrosine kinase activity found to be over-expressed in many tumor types [1].
The crucial role of IGF-1R receptor signaling in malignant transformation and in tumor cell proliferation and survival makes it a very attractive therapeutic target [2–7]. Furthermore, in preclinical models, IGF-1R signaling demonstrated to interfere with numerous receptor pathways and was implicated in the mechanism to render tumor cell resistant to chemotherapy, antihormonal therapy and to anti-EGFR and HER2 targeted therapies [8–21]. Similarly, mammalian target of rapamycin (m-TOR) inhibitors can activate PI3K-Akt pathway via loss of negative feedback on IRS-1 (insulin receptor substrate—1), an effect that can be suppressed by IGF-1R blockade [22–24].
Recently, the anti-neoplastic activity of IGF-1R antibodies became one of the most investigated in clinical oncology. Almost 30 candidate drugs were tested in more than 70 clinical trials conducted in a wide variety of cancers through academia, pharmaceutical and biotechnology companies. Early clinical data suggested the activity of IGF-1R target-drugs in selected tumor types, such as Ewing’s sarcoma, non-small cell lung cancer, adrenocortical carcinoma, but the initial enthusiasm quickly encountered several challenges and disappointment.
In his review we focus on the most relevant clinical data concerning tumor types where IGR-1R targeting was considered of potential interest. Meanwhile, based on existing clinical and translational data this article describes some potential biomarkers that could help to better identify the patient population who would benefit most.
IGF-1R pathway
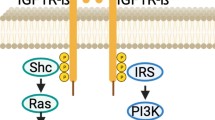
Binding of IGF-1 or IGF-2 to IGF-1R leads to receptor auto-phosphorylation. Ligand bioavailability is elevated in some tissues and is highly dependent on IGF-binding proteins (IGFBP) and IGFBP-proteases. Activation of the receptor leads to the recruitment of multiple adaptor proteins such as insulin receptor substrates (IRS), Shc and Src homologue and subsequent activation of at least two pro-survival signaling pathways. The main event following IGF-1R phosphorylation is the stimulation of phophoinositol 3-kinase (PI3K)-Akt signaling pathway, leading to cell survival. The second pathway consists of Ras, Raf and extracellular-signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) activation, leading to tumor growth and proliferation [4–6]. Figure 1

Downstream signaling of the IGF-1R. IGF-1R is a transmembrane tyrosine-kinase receptor, binding either IGF1 or IGF2. Ligand binding leads to IRS phosphorylation and recruitment of regulatory (p85) and catalytic (p110) subunits of PI3K and subsequently Akt phosphorylation on threonine 308. Serine 473 of Akt is phohoshorylated by mTORC2 complex also activated by IGF-1R by an unknown mechanism. Akt promotes cell survival by multiple mechanisms, inhibiting apoptosis and inducing expression of prosurvival genes. The other parallel pathway, related to IGF-1R by IRS or Shc proteins is the RAS-RAF-MAPK and JNK, resulting in cell proliferation. Negative regulation by mTORC1, S6K, JNK and ERKs induces IRS1 degradation. ( AKT—protein kinase ; ERK—extracellular-signal-regulated kinase; MAPK—mitogen activated protein kinase; Grb2—growth factor receptor-bound protein 2; IGF-1R—insulin-like growth factor 1 receptor; GSK3—glycogen synthase kinase 3; IRS 1—insulin-like receptror substrate 1; MEK—mitogen-activated protein kinase; mTOR—mammalian target of rapamycin, PI3K—phosphatidylinositol 3-kinase; PIP2—phosphatidylinositol 4,5-biphosphate; PIP3—phosphatidylinositol 3,4,5 triphosphate; PTEN—phosphatase and tensin homolog; RHEB—Ras homolog enriched in brain; S6K—S6 kinase; SOS—son of sevenless; TSC2—tuberous sclerosis complex 2)
Although IGF-1R axis components can be highly altered in cancer, little is known about molecular mechanisms involved in this process. Chromosome 15q26, where IGF-1R is located was found to be amplified in basal-like breast cancer [25]. Low levels of IGF-1R copy number gain were also shown in lung cancer [26, 27], pancreatic adenocarcinoma and colon cancer [28–30]. A more recent study has demonstrated that KIT and PDGFR-α wild type GIST have a significantly higher level of IGF-1R amplification than mutated ones [31]. Whereas no mutation of IGF-1R was described to date, there are some reports of gene polymorphism encoding IGF-1 or IGFBP-3.
Several approaches targeting IGF-1R were developed, including monoclonal antibodies, small molecule tyrosine kinase inhibitors and ligand binding antibodies. The most advanced in clinical development are monoclonal antibodies. This approach is considered more selective in blocking IGF-1R activity than tyrosine kinase inhibition. IGF-1R shows high homology with insulin receptor (IR), thus small molecule TKIs may block IR and receptor hybrids as well [2]. This could be an advantage in term of metabolic consequences, considering the high homology between IGF-1R and IR. Selectivity is also important when we take into account treatment efficacy. IR and IR/IGF-1R hybrids represent an important role in IGF signaling, and non-selective TK inhibitors can additionally abrogate their activity. Thus the development of techniques able to differentially assess IGF-1R/IR hybrids and homodimers is necessary to efficiently tailor anti-IGF-1R therapies.
Toxicities reported in phase I trails were uncommon and maximal tolerated dose was not identified for either of the drugs blocking IGF-1R investigated as single agent. Hyperglycemia was more frequently observed with tyrosine kinase inhibitors. Up to 20 % of the patients experienced this toxicity and it was usually mild to moderate and manageable with oral anti-diabetic medication. Other frequently registered toxicities were fatigue (8–14 %), and mild skin toxicities (rash, urticaria and pruritus). Hypersensitivity reaction was a rare event [42, 44, 51, 52]. Hematological side effect was a rare but important when occurred. Grade 3 thrombocytopenia was considered a DLT with 20 mg/kg of AMG-479 and lymphocyte count decrease occurred in 7 % of the patients treated with CP-751, 871 [44, 51].
IGF-1R inhibitors in lung cancer
Insulin growth factor receptor is overexpressed in up to 80 % of lung cancers (1). Pre-clinical data on cell lines demonstrate that IGF-1R activation induces resistance to anti-epidermal growth factor receptor (EGFR) therapy through activation of PI3K-AKT pathway [12–17].
The first retrospective analysis of IGF-1R expression in primary resected NSCLC (n = 184), demonstrated a positive correlation between IGF-1R expression and poor overall survival [32]. The same authors recently concluded that IGF-1R protein expression is significantly higher in squamous cell carcinomas (67 %) when compared with other histologies (24 %). The prognostic value of IGF-1R expression is controversial across different studies [26, 32–34]. As an example IGF-1R gene copy number gain (3 % amplification and 24 % high polysomy) had surprisingly positive prognostic value [26]. High expression level of IGF-1R was related to treatment response in a preclinical study on 22 NSCLC cell lines [26] and in a small cohort of patients treated with figitumomab [26, 27, 35]. The predictive value of IGF-1R copy number gain needs further investigation.
The initial hints of anti-IGF-1R activity in advanced NSCLC observed across several phase I studies were subsequently reproduced in one randomized phase II trial [36]. These trials investigated the efficacy of figitumomab, a selective fully human IgG2 monoclonal antibody against insulin like growth factor receptor 1. A total of 156 patients were allocated to carboplatin, paclitaxel and two dose levels of figitumumab (10 mg/Kg or 20 mg/Kg), or carboplatin paclitaxel alone. At the moment of disease progression, patients who received CT alone were treated with figitumumab with or without other CT regimen (investigator’s discretion). The addition of figitumumab to carboplatin-paclitaxel significantly increased the overall response rate (54 % versus 42 %) [33]. Exploratory analyses revealed a dose-dependent response in patients with squamous-cell histology treated with 20 mg/Kg of figitumumab plus CT with a trend towards better PFS [33].
These encouraging results lead to its subsequent evaluation in phase III trials. Two randomized phase III studies in advanced NSCLC were prematurely discontinued after Independent Data Monitoring Committees analysis indicating that the addition of figitumumab to carboplatin-paclitaxel would be unlikely to meet the primary endpoint of improving overall survival. Additionally more treatment related deaths and serious adverse events were reported in the combination arm. Cardiac toxicity was two times higher in patients treated with figitumomab (3 % versus 1.2 %). High baseline circulating IGF-1 level was predictive of better survival in figitumomab-carboplatin-paclitaxel arm [37]. Unmet primary endpoint could be attributed at least partially to the heterogeneous population of advanced NSCLC included and no available stratification according to hystological subtypes. Also, there was not an exploratory evaluation of potential biomarkers that could be eventually related to treatment response.
Finally, a third planned phase III trials evaluating the combination of figitumumab with cisplatin and gemcitabine (ADVIGO1017) was cancelled prior to study enrollment [38].
It’s somehow unfortunate that the initial encouraging activity of IGF-1R targeting combined with standard chemotherapy was not confirmed in large phase III clinical trials. Careful selection of patient population according to histological subtypes and smart trial design which could lead to the identification of putative predictive biomarkers, beyond circulating IGF-1 levels, might be more successful and permitting less costly drug approval strategies.
IGF-1R inhibitors in sarcoma
IGF-1R axis alteration were described in many sarcoma subtypes. In Ewing sarcoma, characterized by EWS/FLI-1 translocation, enhanced IGF-1R activity has been observed. This was related to the transcriptional repression of the IGFBP-3, increasing IGF-1 ligand bioavailability with resulting IGF-1R activation. In desmoplastic small round cell tumors (DSRCT) EWS-WT1 translocation induces a threefold over-expression of IGF-1R [39, 40]. Gastrointestinal stromal tumors (GISTs) lacking of KIT and PDGFRα mutations presented significantly higher prevalence of IGF-1R amplification compared to mutated ones [31]. High IGF-1 and IGF-2 expression levels were associated to a highly malignant phenotype and negative prognostic value in wild type and mutant GISTs [41]. In addition, increased IGF-2 levels was described in many other sarcoma subtypes, such as rhabdomyosarcomas, leiomyosarcomas and synovial sarcomas, suggesting an autocrine/paracrine dependency on this pathway [41].
Based on the strong biological rational of inhibiting IGF-1R mediated signaling in sarcoma, many clinical trials were conducted or are ongoing in this patient population (Table 1).
The most striking evidence of clinical activity emerges from Ewing sarcoma. The results of two phase II trials were recently published, evaluating the efficacy and safety of R1507 (robatumumab, a fully human IgG1 mAb to IGF-1R) in recurrent and refractory Ewing’s sarcomas and AMG 479 (fully human mAb to IGF-1R) in recurrent refractory Ewing’s family of tumors and desmoplastic small round cell tumors (DSRT). In the SARC 001 study 111 Ewing’s sarcoma patients were treated with R1507, administered intravenously at 9 mg/kg weekly. Overall response rate was 9 % (1 complete response and 9 partial responses according to RECIST criteria) and additional 21 % of patients experiencing unconfirmed partial response or disease stabilization. Thus two patterns of response were identified, 9 % of the patients achieving a robust, durable response for about 25 weeks and 6 % having short lived responses. Median progression free survival in this study was 5.7 months and overall survival 6.9 months [42].
Based on the encouraging phase I result with AMG 479 showing a complete response in one Ewing’s sarcoma patients sustained after more than 3 years and a second unconfirmed PR, a phase II trial was conducted in a population of 38 patients having a recurrent or refractory Ewing’s family of tumors (EFT) or DSRCT. Additionally a biomarker analysis was performed, exploring the relation between EWS translocation and clinical response. Two patients (one EFT and one DSCRT) achieved a partial response and almost half of overall patient population had a stable disease. Clinical benefit rate (overall response and disease stabilization for more than 24 weeks) was 17 %. PFS was about 8 weeks for EFT and 19 weeks for DSCRT. Two best responses had predominantly EWS-FLI1 type 2 transcripts, but globally no correlation could be identified between a specific EWS translocation and clinical benefit [43].
Twenty-nine patients with Ewing’s sarcoma and a heterogeneous group of other sarcoma subtypes were treated with single agent figitumumab (CP-751, 871, Pfizer, IgG2 monoclonal antibody to IGF-1R) using a dose of 20 mg/kg every 3 weeks. Although primary endpoints were safety and tolerability, preliminary data of antitumor activity were also provided. Twenty-two patients were evaluable for response and half of them presented tumor shrinkage. One Ewing’s sarcoma patients achieved a pathological complete response and one a partial response, five additional patients having some degree of tumor reduction but remaining in the category of stable disease according to RECIST criteria lasting between 4 and 16 months. Disease stabilization for 4 months or longer was also noticed in one patient having a recurrent synovial sarcoma and an additional one with fibrosarcoma [44].
A phase II single arm study of figitumumab in Ewing’s sarcoma is completing accrual with approximately 130 patients [45]. A phase II trial investigating the efficacy of SCH-717454 (robatumumab, a fully human neutralizing anti IGF-1R antibody) has planned to include 190 patients with osteosarcoma and Ewing’s sarcoma family of tumors [46]. A second trial with cixutumumab (fully human IgG1 moAb) is recruiting 185 patients in 5 arms with different sarcoma subtypes [47].
It can be concluded that monoclonal antibodies targeting IGF-1R produced some activity in sarcoma patients. The major challenge is how to select these patients and what are the best predictive biomarkers of response to these therapies.
IGF—1R inhibitors in breast cancer
IGF-1R overexpression was observed in 44 % of breast cancer tissue specimens, showing no correlation with prognosis [48]. Circulating IGF-1 levels were associated with primary breast cancer risk. This seems to be confined to estrogen-receptor positive tumors and to be not significantly modified by IGFBP-3 levels or menopausal status [49]. High IGF-1 activation was also associated with poor prognosis in breast cancer. IGF-1 gene signature appeared to be up regulated in basal like (ER and HER2 negative) and most of the luminal-B tumors (ER positive but highly proliferative disease) [50].
There is extensive preclinical evidence supporting the synergistic growth inhibition property of combined IGF-1R and HER2 targeting treatment [18, 20, 21].
Increased IGF-1R expression was highly associated with ER status, encoded by estrogen receptor alpha (ESR1) gene. Reciprocal inhibition of ERS1 or IGF-1R transcript levels was produced by siRNA knockdown of one or the other of these targets. Furthermore it was shown in vitro and in vivo synergism of dual targeting of these pathways by fulvestrant or tamoxifen combined with h10H5, an IGF-1R monoclonal antibody [29].
Increased IGF-1R signaling has been also implicated in trastuzumab resistance. A bidirectional cross talk was detected between the two receptors in preclinical studies. Recombinant human IGFBP-3 showed significant inhibition of tumor growth in trastuzumab resistant HER2 and IGF-1R overexpressing cell lines and in vivo synergistic interaction with antiHER2 therapy by decreasing bioavailability of IGF-1 ligand [11, 19].
A physical interaction between IGF-1R and HER2 was found in trastuzumab resistant cells since the phosphorylation of both receptors was stimulated by IGF-1 [10]. In another study both receptors were found in an immunoprecitable complex [18]. HER2 heterodimerisation with other members of HER family is a well-known phenomenon. Besides this, heterodimers with IGF-1R were also described in trastuzumab resistant cells. Another mechanism contributing to trastuzumab resistance is p27kip down-regulation and this was stimulated by IGF-1 in some preclinical models [10].
Several phase I trials assessing the safety of IGF-1R targeted agents demonstrated clinical activity in two advanced breast cancer patients receiving AVE 1642 and one treated by AMG 479 as single agent [51, 52].
Ongoing trials in advanced breast cancer evaluate the activity of different drug combinations with IGF-1R inhibitors. Based on the fact that IGF-1 is up regulated in poor prognosis, ER positive luminal B tumors, Neo-BIG designed a neoadjuvant trial combining letrozole with MK-0646 (BIG 1–09). Unfortunately the clinical development of this protocol was temporary suspended. Additional phase II trials are evaluating the IGF-1R inhibitors associated to endocrine treatment or HER2 inhibitors in advanced breast cancer tumors (Table 2).
Preclinical studies suggest that mTOR inhibitors are able to up-regulate PI3K-Akt pathway by the release of the negative feedback of S6K on IRS-1 [22, 24]. Remarkable activity was seen in breast cancer patients in a phase I dose finding study of oral mTOR inhibitor ridaforolimus associated to IGF-1R monoclonal antibody, MK-0646 (dalotuzumab). Ten out of 23 patients (43 %) diagnosed with metastatic breast cancer experienced clinical activity in the expansion cohort of this study. Most of them had hormone receptor positive tumors with high proliferation rate, defined by Ki67 levels above 15 %. In this specific patient population the response rate was as high as 54 %. Based on these encouraging results a phase II study is ongoing comparing exemestane with the association of ridaforolimus and dalotozumab in HR overexpressing, HER2 negative tumors, failing 1–2 hormonal agents and maximum one chemotherapy regimen for metastatic disease [58]. Surprisingly these results were not reproduced in nine breast cancer patients included in another phase I trial combining Temsirolimus with IMC-A12 (cixitumomab, fully human IgG1 monoclonal antibody). Only one patient with breast cancer had disease stabilization in this study [59]. Overall these data are still immature and unfortunately none of these trials was designed to evaluate in parallel molecular characteristics of individual tumors that could predict eventually treatment response or resistance.
We can conclude that the main area of interest of using IGF-1R targeted agents in breast cancer is a combination strategy with endocrine treatment, HER2 and mTOR targeted agents. Clinical confirmation is in progress.
IGF-1R inhibitors in adrenocortical carcinoma (ACC)
Metastatic and non-surgically manageable adrenocortical carcinoma (ACC) has the reputation of highly resistant disease to classical systemic therapeutic interventions. Some preclinical data suggests that IGF system has an important role in ACC pathogenesis. The vast majority of human ACC specimens display a relevant overexpression of IGF-2 transcripts compared to normal adrenal tissue. This was related to several genetic alterations such as loss of imprinting or loss of heterozygosity of the 11p15 gene locus [60, 61]. Additionally, a high overexpression of IGF-1R was found in ACC samples with concomitant downstream Akt activation, demonstrating the critical role of the pathway in the pathogenesis of this disease [61].
Preliminary antitumor activity was seen in one ACC patient treated with OSI-906 a small-molecule IGF-1R tyrosine kinase inhibitor in a phase I dose-escalation study. A randomized, double blind placebo controlled study is currently including participants evaluating the efficacy of this drug in locally advanced or metastatic ACC [62].
Fourteen ACC patients were treated in the phase I dose expansion cohort with figitumumab. The results of this study are somehow less encouraging, since no relevant clinical activity was demonstrated. However four patients experienced tumor shrinkage without meeting RECIST criteria of partial response [63].
Conclusions and future directions
Clinical validation of IGF-1R as a target emerged with early evidence of activity, especially in Ewing’s sarcoma and other sarcoma subtypes, ACC and NSCLC. However evidence is still limited to draw a definitive and firm conclusion on which patient population may have benefit. Thus, despite the overwhelming early enthusiasm, the development of IGF-1R targeted agents arrived to an important crossroad.
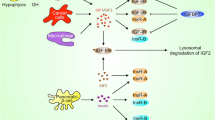
Up to now there are few, if any reliable biomarkers predicting response to IGF-1R targeting agents. Alterations of different IGF-1R axis components were potentially related to efficacy in preclinical studies. (Table 3) Aberrant expression of IGF-1R is detected in many cancers. Some preclinical studies conclude that IGF-1R expression is necessary but not sufficient to predict sensitivity. While phospho-IGF-1R levels seem to not correlate with in vitro effectiveness of IGF-1R targeting, total expression of IGF-1R was a better predictor of response [27, 29]. The number of IGF-1R per cell was correlated with response in some preclinical studies suggesting that somewhere between 1300 and 10000 receptors are necessary to obtain effective cell growth inhibition. These data should be interpreted with caution for many reasons. IGF-1R takes part of a complex autocrine loop involving also its ligands, IGF-1 and IGF-2, as well as intracellular adaptor proteins such as IRS-1 and IRS-2. Meanwhile ligand bioavailability is dependent on IGFBP overexpression. (Figure 2) To give an example: sarcoma cell lines resistant to BMS 536924 (small molecule IGF-1R tyrosine kinase inhibitor) expressed high levels of IGFBP-3 and IGFBP-6 whereas the sensitive ones showed high expression level of IGF-1 and IGF-2 [16]. Other investigators have reported IRS-1 and IRS-2 as predictors of response, suggesting the importance of IGF-1R axis activation in therapeutic response as well [29]. Besides IGF-1R the ligands may also activate IR and IGF-1R/IR hybrids. Generally tyrosine - kinase inhibitors are not selective for IR or IGF-1R, while monoclonal antibodies are blocking only IGF-1R and IGF-1R/IR hybrids. Thus the individual assessment of both receptors and their conformation could have an impact on response to different targeting strategies. One can also speculate that IGF-1R blockage by a therapeutic antibody leads to increased ligand availability to insulin receptor thus conducting to its compensatory activation. The role of the latter in human cancer development is frequently evoked.

Insulin like growth factor (IGF) and insulin receptor (IR) signaling system. The availability of IGF-1 and IGF-2 ligands is highly influenced by IGFBPs (IGF-binding proteins), whereas insulin has direct access to its receptor. IGF-2 can also be sequestered by IGF-2R, which does not activate downstream signaling. Tyrosine-kinase receptors, such as holo-IR, IGF-1R/IR hybrids and holo-IGF-1R phosphorylate their adaptor proteins (IRSs), by this way conducting to downstream receptor signaling activation. Phosphoinsitide 3-kinase (PI3K)—Akt—mammailian target of rapamycin (m-TOR) is a critical pathway in IGF-1R signaling. ( IGF1, 2—insulin-like growth factor 1 and 2, IGFBPs—IGF binding proteins, IR—insulin receptor, IGF-1R—insulin-like growth factor receptor-1, IGF-2R—insulin-like growth factor receptor-2, IRS—insulin receptor substrate)
No activating mutations of IGF-1R have been described so far. On the other hand IGF-1R gene copy number gain was identified in some tumor types, such as wild-type GIST, breast cancer and NSCLC. This might support the idea that IGF-1R gene amplification and its relationship to treatment response is worthy to be evaluated.
IGF-1R phosphorylation leads to the activation of multiple signaling pathways. The antiapoptotic phosphoinositol 3-kinase (PI3K) -Akt pathway is a critical pathway in IGF-1 signaling, although it can mediate also signal from other growth factor receptors. Downstream signaling inhibition could be not only a useful pharmacodynamic biomarker, but also its examination before and after treatment could be informative regarding treatment response as well.
Taking into account the above-mentioned data, clinically quantifiable biomarkers should be developed, promoting smarter trial design to select patient population treated with IGF-1R targeted agents. Identification of molecular abnormalities of IGF-1R as well as at the level of downstream pathway components or taking IGF-1R as a part of a complex autocrine loop would be some examples from preclinical data worthy to be translated into the clinics.
Another issue that might explain, at least partly the concern regarding initial clinical results is that efficacy of IGF-1R targeting as a single agent is a rare event. Thus development of rational combinations with other anticancer agents needs to be explored. Parallel inhibition of a second growth factor receptor such as EGFR or HER2 or a concomitant downstream signaling blockade such as m-TOR or PI3K could considerably enhance antitumor activity. Combination of two HER-2 targeting antibodies directed against different epitopes of Cerb-B2 receptor showed improvement of clinical activity in the treatment of HER-2 amplified metastatic breast cancer. Pertuzumab inhibits HER-2 dimerization by preventing its pairing with other members of HER receptors. Combined treatment with trastuzumab and pertuzumab proved high efficacy in trastuzumab resistant metastatic breast cancer as well in the neo-adjuvant setting [64, 65]. Dual targeting of IGF-1R by two antibodies with distinct mechanism of action producing in the mean time an allosteric and a competitive blockage showed promising preclinical activity in one study [66, 67].
In conclusion the clinical development of IGF-1R targeted agents should be carefully reassessed while placing upfront the understanding and the identification of molecular markers predicting treatment sensitivity and resistance and the investigation of combination therapeutic strategies. In addition it is of utmost importance to optimally design clinical trials.
References
Ouban A, Muraca P, Yeatman T et al (2003) Expression and Distribution of Insulin-Like Growth Factor-1 Receptor in Human Carcinomas. Hum Pathol 34:803–808
Gualberto A, Pollak M (2009) Emerging role of insulin-like growth factor receptor inhibitors in oncology: early clinicl trial results and future directions. Oncogene 28:1–13
Valentinis B, Baserga R (2001) IGF-I receptor signalling in transformation and differentiation. J Clin Pathol: Mol Pathol 54:133–137
Baserga R, Peruzzi F, Reiss K (2003) The IGF-1 Receptor in Cancer Biology. Int J Cancer 107:873–877
Samani AA, Yakar S, LeRoith D et al (2007) The Role of the IGF System in Cancer Growth and Metastasis: Overview and Recent Insights. Endocr Rev 28:20–47
Chitnis MM, Yuen SP, Protheroe SA et al (2008) The Type 1 Insulin-Like Growth Factor Receptor Pathway. Clin Cancer Res 14:6364–6370
Pollak M (2008) Insulin and insulin-like growth factor signaling in neoplasia. Nat Rev Cancer 8:915–928
Dunn SE, Hardman RA, Kari FW et al (1997) Insulin-like Growth Factor 1 (IGF-1) Alters Sensitivity of HBL100 Human Breast Cancer Cells by Inhibition of Apoptosis Induced by Divers Anticancer drugs. Cancer Res 57:2687–2693
Eckstein N, Servan K, Hildebrand B, Politz A et al (2009) Hyperactivation of the Insulin-like Growth Factor Receptor I Signalling Pathway Is an Essential Event for Cisplatin Resistance of Ovarian Cancer Cells. Cancer Res 69:2296–3003
Natha R, Yuan LX, Zhang B et al (2005) Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerisation contributes to trastuzumab resistance of breast cancer cells. Cancer Res 65:11118–11128
Lu Y, Zi X, Zhao Y et al (2001) Insulin-like growth factor-I receptor signalling and resistance to trastuzumab (Herceptin). J Natl Cancer Inst 93:1852–1857
Knowlden JM, Jones HE, Barrow D et al (2008) Insulin receptor substrate-1 involvement in epidermal growth factor receptor and insulin—like growth factor receptor signaling: implication for Gefitinib (‘Iressa’) response and resistance. Breast Cancer Res Treat 111:79–91
Jones HE, Goddard L, Gee GM et al (2004) Insulin-like growth factor I receptor signalling and acquired resistance to gefitinib (ZD1839; Iressa) in human breast and prostate cancer cells. Endocr Relat Cancer 11:793–814
Chakravarti A, Loeffler JS, Dyson NJ (2002) Insulin-like Growth Factor Receptor I Mediates Resistance to Anti-Epidermal Growth Factor Receptor Therapy in Primary Human Glioblastoma Cells through Continued Activation of Phosphoinositide 3 -Kinase Signaling. Cancer Res 62:200–207
Buck E, Eyzaguirre A, Rosenfeld-Franklin M et al (2008) Feedback Mechanisms Promote Coopeativity for Small Molecule Inhibitors of Epidermal and Insulin-Like Growth Factor Receptors. Cancer Res 68:8322–8332
Huang F, Greer A, Hurlburt W et al (2009) The Mechanisms of Differential Sensitivity to an Insulin-like Growth Factor-1 Receptor Inhibitor (BMS-536924) and Rationale for Combining with EGFR/HER2 Inhibitors. Cancer Res 69:161–170
Hu PY, Patil BS, Panasiewitz M et al (2008) Heterogeneity of receptor Function in Colon Carcinoma Cells Determined by Cross-talk between Type I Insulin-like Growth Factor Receptor and Epidermal Growth Factor Receptor. Cancer Res 68:8004–8013
Chakraborty AK, Ke L, DiGiovanna MP (2008) Co-Targeting Insulin-Like Growth Factor I Receptor and Her2: Dramatic Effects of HER2 Inhibitors on Nonoverexpressing Breast Cancer. Cancer Res 68:1538–1545
Jin Q, Esteva FJ (2008) Cross Talk Between the ErbB/HER Family and Type I Insulin-Like Growth Factor Receptor Signalling Pathway in Breast Cancer. J Mammary Gland Biol Neoplasia 13:485–498
Haluska P, Carboni JM, TenEyck C et al (2008) HER receptor signalling confers resistance to the insuline-like growth factor-I receptor inhibitor, BMS-536924. Mol Cancer Ther 7:2589–2608
Browne BC, Crown J, Venkatesan N et al (2011) Inhibition of IGF1R activity enhances response to trastuzumab in HER-2 positive breast cancer cells. Ann Oncol 22:68–73
Wan X, Harkavy B, Shen N et al (2007) Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 26:1932–1940
Haruta T, Uno T, Kawahara J et al (2009) A Rapamycin-Sensitive Pathway Down-Regulates Insulin Signalling via Phosphorylation and Proteasomal Degradation of Insulin Receptor Substrate-1. Molecular Endocrinol 14:783–794
Takano A, Usui I, Haruta T et al (2001) Mammalian Target of Rapamycin Pathway Regulates Insulin Signalling via Subcelular Redistribution of Insulin Receptor Substrate 1 and Integrates Nutritional Signals and Metabolic Signals of Insulin. Mol an Cel Biol 21:5050–5062
Adélaide J, Finetti P, Bekhouche I et al (2007) Integrated Profiling of Basal and Luminal Breast Cancers. Cancer Res 67:11565–11575
Dziadziuszko R, Merrick DT, Samir E (2010) Insulin-like Growth Factor Receptor I (IGF1R) Gene Copy Number Is Associated With Survival in Operable Non-Small-Cell Lung Cancer: A Comparison Between IGF1R Fluorescent In Situ Hybridization, Protein Expression, and mRNA Expression. J Clin Oncol 28:2174–2180
Gong Y, Yao E, Shen R et al (2009) High Expression Levels of Total IGF-1R and Sensitivity of NSCLC Cells In Vitro to an Anti-IGF-1R Antibody (R1507). PLoS One 4:e7273
Armengol G, Knuutila S, Lluis F et al (2000) DNA copy number changes and evaluation of MYC, IGF1R, and FES amplification in xenografts of pancreatic Aden carcinoma. Cancer Genet Cytogenet 116:133–141
Zha J, O’Brien C, Savage H et al (2009) Molecular predictors of response to a humanized anti-insulin-like growth factor-I receptor monoclonal antibody in breast and colorectal cancer. Mol Cancer Ther 8:2110–2121
Pitts TM, Tan AC, Kulikowski GN et al (2010) Development of an Integrated Genomic Classifier for a Novel Agent in Colorectal Cancer: Approach to Individualized Therapy in Early Development. Clin Cancer Res 16:3193–3204
Tarn C, Rink L, Flieder D et al (2008) Insulin-like growth factor receptor is a potential therapeutic target for gastrointestinal stromal tumors. PNAS 24:8387–8392
Merrick DT, Dziadziuszko R, Szostakiewicz B, Szymanowska A, Rzyman W, Jassem E et al (2007) High insulin-like growth factor 1 receptor (IGF1R) expression is associated with poor survival in surgically treated non-small cell lung cancer (NSCLC) patients (pts). J Clin Oncol 25 (18_suppl):Abst 7550.
Ludovini V, Bellezza G, Pistola L, Bianconi F, Di Carlo L, Sidoni A et al (2009) High coexpression of both insulin-like growth factor receptor-1 (IGFR-1) and epidermal growth factor receptor (EGFR) is associated with shorter disease-free survival in resected non-small-cell lung cancer patients. Ann Oncol 20:842–849
Cappuzzo F, Tallini G, Finocchiaro G et al (2006) Insulin-like growth factor receptor & (IGFR-1) is significantly associated with longer survival in non-small cell lung cancer (NSCLC) patients. Ann Oncol 17:1120–1127
Gualberto A, Dolled-Filhart M, Gustavson M et al (2010) Molecular Analysis of Non-Small Cell Lung Cancer Identifies Subsets with Different Sensitivity to Insulin-like Growth Factor I Receptor Inhibition. Clin Cancer Res 16:4654–4665
Karp DD, Paz-Ares LG, Novello S, Haluska P, Garland L, Cardenal F et al (2009) Phase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751, 871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancer. J Clin Oncol 27:2516–2522
Jassem J, Langer CJ, Karp DD et al (2010) Randomized, open label, phase III trial of figitumomab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin in patients with non-small cell lung cancer. J Clin Oncol 28 (15 suppl): Abstr 7500
Study Of The Effect Of CP-751,871 In Combination With Gemcitabine And Cisplatin In Patients With Advanced Non-Small Cell Lung Cancer (ADVIGO1017) http://clinicaltrials.gov/ct2/show/NCT00907504. Accessed 15 April 2011
Rikhof B, de Jong S, Suurmeijer AJH et al (2009) The insulin-like growth factor system and sarcomas. J Pathol 217:469–482
Olmos D, Tan DS, Jones RL et al (2010) Biological Rationale and Current Clinical Experience With Anti-Insulin-Like Growth Factor 1 Receptor Monoclonal Antibodies in Treating Sarcoma. The Cancer Journal 16:183–194
Braconi C, Bracci R, Bearzi I et al (2008) Insulin-like growth factor (IGF) 1 and 2 help to predict disease outcome in GIST patients. Ann Oncol 19:1293–1298
Pappo AS, Patel S, Crowley J et al (2010) Activity of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF1R), in patients with recurrent or refractory Ewing’s sarcoma family of tumors (ESFT): Results of a phase II SARCC study. J Clin Oncol 28: 15s, 2010 (suppl; abstr 10000)
Tap WD, Demetri GD, Barnette P et al. AMG 479 in relapsed or refractory Ewing’s family tumors (EFT) or desmoplastic small round cell tumors (DSCRCT): Phase II results. J Clin Oncol 28 (15 suppl): Abstr 10001
Olmos D, Postel-Vinay S, Molife LR et al (2010) Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase I expansion cohort study. Lancet Oncol 11:129–135
Study Of CP-751,871 In Patients With Ewing’s Sarcoma Family Of Tumors http://clinicaltrials.gov/ct2/show/NCT00560235?term=CP-751%2C871+sarcoma&rank=2. Accessed 20 April 2011
A Study to Determine the Activity of SCH 717454 in Subjects with Relapsed Osteosarcoma or Ewing's Sarcoma (Study P04720AM3) http://clinicaltrials.gov/ct2/show/NCT00617890?term=SCH-717454&rank=2. Accessed 20 April 2011
A Five-Tier Open Label Study of IMC-A12 in advanced Sarcoma http://clinicaltrials.gov/ct2/show/NCT00668148?term=cixutumumab+sarcoma&rank=2. Accessed 20 April 2011
Shimizu S, Hasegawa T, Tani Y et al (2005) Expression of insulin-like growth factor 1 receptor in primary breast cancer: Immunohistochemical analysis. Human Pathol 36:448–449
Key TJ, Appleby PN, Reeves GK et al (2010) Insulin-like growth factor 1 (IGF1), IGF binding protein 3 (IGFBP3), and breast cancer risk: pooled individual data analysis of 17 prospective studies. Lancet Oncol 11:530–542
Creighton CJ, Casa A, Lazard Z et al (2008) Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol 26:4078–4085
Tolcher AW, Ptnaik A, Till E et al (2008) A phase I study of AVE1642, a humanized monoclonal antibody IGF-1R (insulin like growth factor 1 receptor) antagonist, in patients (pts) with advanced solid tumors (ST). J Clin Oncol 26 (May 20 suppl): Abstr 3582
Tolcher AW, Rothenberg ML, Rodon J et al (2007) A phase I pharmacokinetic and pharmacodynamic study of AMG 479, a fully human monoclonal antibody against insulin-like growth factor type 1 receptor (IGF-1R), in advanced solid tumors. J Clin Oncol 25 (June 20 Suppl): Abstr 3002
Study Of CP-751,871 In Combination With Exemestane In Postmenopausal Women With Hormone Receptor Positive Advanced Breast Cancer http://clinicaltrials.gov/ct2/show/NCT00372996. Accessed 20 April 2011
A Study for Safety and Effectiveness of IMCA12 by Itself or Combined With Antiestrogens to Treat Breast Cancer http://clinicaltrials.gov/ct2/show/NCT00728949?term=NCT00728949&rank=1. Accessed 20 April 2011
Capecitabine and Lapatinib With or Without Cixutumumab in Treating Patients With Previously Treated HER2-Positive Stage IIIB, Stage IIIC, or Stage IV Breast Cancer http://clinicaltrials.gov/ct2/show/NCT00684983?term=NCT00684983&rank=1 Accessed 20 April 2011
A Study of AMG 479 With Exemestane or Fulvestrant in Postmenopausal Women With Hormone Receptor Positive Locally Advanced or Metastatic Breast Cancer http://clinicaltrials.gov/ct2/show/NCT00626106?term=NCT00626106&rank=1. Accessed 20 April 2011
Combination Study of BMS-754807 and Herceptin® in Patients With Advanced or Metastatic Her-2-positive Breast Cancer http://clinicaltrials.gov/ct2/show/NCT00788333?term=NCT00788333&rank=1. Accessed 20 April 2011
Di Cosimo S, Bendell JC, Cervantes-Ruiperez A et al (2010) A phase I study of the oral mTOR inhibitor ridaforolimus (RIDA) in combination with the IGF-1R antibody dalotozumab (DALO) in patients (pts) with advanced solid tumors. J Clin Oncol 28 (15 suppl): Abstr 3008
Naing A, LoRusso P, Gupta S et al (2010) Dual inhibition of IGFR and mTOR pathways. J Clin Oncol 28 (15 suppl): Abstr 3007
West AN, Neale GA, Pounds S et al (2007) Gene Expression Profiling of Childhood Adrenocortical Tumors. Cancer Res 67:600–608
Barlaskar FM, Spalding AC, Heaton JH et al (2009) Preclinical Targeting of the Type I Insulin-Like Growth Factor Receptor in Adrenocortical Carcinoma. J Clin Endocrinol Metab 94:204–212
Carden CP, Frentzas S, Langham M et al (2009) Preliminary activity in adrenocortical tumor (ACC) in phase I dose escalation study of intermittent oral dosing of OSI-906, a small-molecule insulin-like growth factor-1 receptor (IGF-1R) tyrosine kinase inhibitor in patients with advanced solid tumors. J Clin Oncol 27 (15 suppl): Abstr 3544
Haluska P, Worden F, Olmos D et al (2010) Safety, tolerability, and pharmacokinetics of the anti-IGF-1R monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma. Cancer Chemother Pharmacol 65:765–773
Baselga J, Gelmon KA, Verma S et al (2010) Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol 28:1138–1144
Gianni L, Pienkowski T, Im Y-H et al. Neaoadjuvant Pertuzumab (P) and Trastuzumab (H): Antitumor and Safety Analysis of a Randomized Phase II Study (‘NeoSphere’). Presented at the 33rd Annual San Antonio Breast Cancer Symposium, December 8–12, 2010. Available online at http://www.abstracts2view.com/sabcs10/view.php?nu=SABCS10L_291&terms= (Accessed 01 Mai 2011).
Dong J, Demarest SJ, Sereno A et al (2010) Combination of Two Insulin-Like Growth Factor-I Receptor Inhibitory Antibodies Targeting Distinct Epitopes Leads to an Enhanced Antitumor Response. Mol Cancer Ther 9:2693–604
Olmos D, Basu B, de Bono J (2010) Targeting Insulin-Like Growth Factor Signaling: Rational Combination Strategies. Mol Cancer Ther 9:2447–2449
Conflict of Interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gombos, A., Metzger-Filho, O., Dal Lago, L. et al. Clinical development of insulin-like growth factor receptor—1 (IGF-1R) inhibitors: At the crossroad?. Invest New Drugs 30, 2433–2442 (2012). https://doi.org/10.1007/s10637-012-9811-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-012-9811-0