
Abstract
Background
AT-rich interactive domain 1A (ARID1A) is a tumor suppressor gene that is frequently mutated in gastric cancer (GC). Although ARID1A mutations are not a druggable target for conventional treatments, novel therapeutic strategies based on a synthetic lethal approach are effective for ARID1A-deficient cancers. The histone methyltransferase EZH2 acts in a synthetic lethal manner in ARID1A-mutated ovarian cancer, although its role in GC remains unknown.
Methods
The selective sensitivity of the EZH2 inhibitors for ARID1A-deficient GC cells was evaluated using cell viability and colony formation assays. The expression of PI3K/AKT signaling genes were investigated using TCGA’s cBioPortal database to determine whether the homeostasis between ARID1A and EZH2 is related to cell proliferation and survival via the PI3K/AKT signaling pathway. We also evaluated the phosphorylation of PI3K/AKT signaling proteins in ARID1A knock downed ARID1A-WT GC cells.
Results
EZH2 inhibitors decreased the viability of ARID1A-deficient cells in a dose-dependent manner and demonstrated the selective sensitivity to ARID1A-deficient cells in vitro experiment system. Bioinformatics approach revealed that the PI3K/AKT signaling was tended to be activated in ARID1A-deficient GC enhancing cell viability and, furthermore, down-regulation of EZH2 in ARID1A-deficient GC was related to normalization of PI3K/AKT signaling pathway. The cell experiment revealed that phosphorylated AKT was upregulated in ARID1A-deficent GC cells.
Conclusions
The present findings provide a rationale for the selective sensitivity of EZH2 inhibitors against ARID1A-deficient GC and suggest the potential efficacy of targeted therapy using EZH2 inhibitors in this patient population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastric cancer (GC) is a leading cause of cancer related morbidity and mortality worldwide, especially in Asia [1, 2]. Despite advances in surgical technique and improvement in the efficacy of chemotherapy, the prognosis of patients with GC remains poor in advanced stage or recurrence [1]. A recent large-scale genome sequencing study from The Cancer Genome Atlas (TCGA) provided important information on the genomic basis and heterogeneity of GC [3]. This led to the identification of several actionable and druggable targets in GC, such as human epidermal growth factor receptor 2 (HER2) amplification, fibroblast growth factor receptor 2 (FGFR2) amplification/mutation, programmed cell death ligand 1 and 2 (PD-L1 and PD-L2) overexpression (CD274 and PDCD1LG2 amplifications) [4]. Mutation profiling demonstrated the potential of personalized therapy based on these genomic aberrations for the treatment of GC. However, targeted therapies need to be further developed to improve their efficacy for the treatment of GC.
AT-rich interactive domain 1A (ARID1A) is a subunit of the Switch/Sucrose Non-fermentable (SWI/SNF) chromatin remodeling complex, which modulates gene expression (5, 6). ARID1A is a haploinsufficient tumor suppressor gene and acts as a driver gene for GC development [7, 8]. Most ARID1A mutations in GC are truncating mutations, leading to ARID1A protein loss; therefore, ARID1A mutations are not considered as a druggable target for conventional therapies [3]. A novel therapeutic approach based on the vulnerability of tumor cells harboring loss-of-function mutations to inhibition of synthetic lethal targets could be effective [9]. Synthetic lethality is generally defined by an interdependent relationship between two genes in which deficiency in gene A or B does not lead to cell death, whereas simultaneous deficiency of both A and B leads to cell death [10]. One example of therapeutic strategy based on synthetic lethality is poly (ADP)-ribose polymerase (PARP)1-targeted therapy against hereditary breast and ovarian cancers harboring BRCA1 and BRCA2 mutations [9]. In ARID1A-deficient cancer, several synthetic lethal targets were identified including enhancer of zeste homolog 2 (EZH2), ARID1B, PARP, ataxia-telangiectasia-mutated-and-Rad3-related kinase (ATR), histone deacetylase 6 (HDAC6) and the glutathione metabolic pathway [10,11,12,13,14,15].
EZH2, a histone methyltransferase and a member of polycomb repressive complex 2 (PRC2), catalyzes the trimethylation of lysine 27 on histone H3; activated EZH2 promotes cancer development by altering the expression of lineage specification genes, the pRB tumor suppressor, or DNA-damage repair genes [16]. Therefore, enzymatic inhibition of EZH2 methyltransferase activity is an effective targeted therapy for cancers with EZH2 overexpression or gain of function mutations [17,18,19]. EZH2 inhibition is also effective for ARID1A-mutated ovarian cancer [12]. ARID1A-deficient cancers are dependent on a non-catalytic role of EZH2, suggesting partial dependence on EZH2 histone methyltransferase activity, but promising therapeutic utility for ARID1A-deficient cancer [20]. A previous study revealed that phosphoinositide-3-kinase interacting protein 1 (PIK3IP1) as a direct target of ARID1A and EZH2, contributing to the selective sensitivity of EZH2 inhibitor for ARID1A-mutated ovarian cancer cells by inhibiting phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT) signaling [12]. However, the therapeutic utility and molecular mechanism underlying synthetic lethality of EZH2 inhibitors for ARID1A-deficient GC remains unknown.
In the present study, we investigated the selectivity of EZH2 inhibitors for ARID1A-deficient GC cells to develop an effective targeted therapeutic strategy by using an in vitro experiment system and bioinformatics approach with the public database. Additionally, we evaluated the candidate group who are beneficial for effect of EZH2 inhibitors on ARID1A-deficient GC patients in association with the clinicopathological characteristics of patients.
Materials and methods
Cell culture
GC cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA) and RIKEN BioResource Center (Ibaraki, Japan). These cells lines were authenticated by short tandem repeat analysis in April 2020 (Promega) [21]. Cells were cultured in RPMI-1640 medium (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Monolayer cells were maintained in a 37 °C incubator with 5% CO2 and observed regularly under a light microscope (magnification, 40 ×); cells were subcultured when they reached 80–90% confluency. All experiments were performed in triplicate. All experiments were carried out in accordance with the approved study plan and relevant guidelines.
Colony formation assay
In this study, three kinds of EZH2 inhibitors, GSK126 (IC50 9.9 nM), GSK343 (IC50 4 nM), and EPZ005687 (IC50 54 nM) were used (Selleck Chemicals, Houston, TX, USA). The effect of drug treatment on cancer cell survival was evaluated using colony formation assays. Cells were trypsinized, counted, reseeded at the specified density in 6-well plates, exposed to three EZH2 inhibitors for 10–14 days, and fixed for 10 min in 50% (v/v) methanol containing 0.01% (w/v) crystal violet. The maximum effective concentration of each drug was determined in previous studies as follows; 5 μM GSK126 [12], 5 μM GSK343 [22], and 8.3 μM EPZ005687 [17]. The number of colonies were counted in all field of each well by OpenCFU (https://opencfu.sourceforge.net/) [23].
Cell proliferation assay
Cell proliferation was evaluated using the WST-cell proliferation assay with the Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the manufacturer’s specifications. Cells were seeded in 96-well plates at density of 1 × 104 cells/well 2 days after small interfering RNA (siRNA)-mediated knockdown of ARID1A and incubated at 37 °C. Different doses of EZH2 inhibitors were added when most cells had attached to the well. After further incubation for 24 h, the cells were treated with 10 µL CCK-8 reagent, incubated at 37 °C for 4 h, and the absorbance at 450 nm was measured a microplate reader. The experiments were independently performed at least three times.
Small interfering RNA transfection
The method used for siRNA transfection was described in the previous study [5]. Briefly, knockdown experiments were performed using siRNA oligos for ARID1A (s15784 and s15785; Thermo Fisher Scientific Inc., Waltham, MA, USA), including two target-specific siRNAs and a control siRNA (negative control #1; Thermo Fisher Scientific) according to the manufacturer’s protocol. During the exponential growth phase, cells were transiently transfected with siRNA at a final concentration of 5 nM using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s protocol. After 48 h of incubation, cells were used for each experiment.
Western blotting
Western blot analysis was performed as previously described [5]. Cells were washed twice in ice-cold PBS, pelleted by centrifugation (1500 rpm for 5 min) and stored at − 80 °C. The pellet was resuspended in radioimmunoprecipitation assay buffer (Thermo Fisher Scientific) with a Halt Protease Inhibitor Single-Use Cocktail (100 ×; Thermo Fisher Scientific), and centrifuged at 4 °C and 15,000 rpm for 20 min. Total protein concentration was measured using the Bradford method (Bio-Rad Laboratories Inc., Hercules, CA, USA) and a Smart Spec 3000 spectrometer (Bio-Rad Laboratories). Tris–Glycine SDS sample buffer (Thermo Fisher Scientific) and 3-Mercapto-1,2-propandiol (Wako Pure Chemical Industries, Ltd., Osaka, Japan) were added to the total protein samples and heated at 100 °C for 3 min. The Tris–Glycine gels (4–20%, Thermo Fisher Scientific) were loaded with 20 µg of protein samples and electrophoresed at 125 V for 40 min using Tris–Glycine SDS running buffer in an Invitrogen™ XCell SureLock™ electrophoresis system (Thermo Fisher Scientific). Then, proteins were transferred onto PVDF membranes using the iBlot 2 Dry Blotting System (Thermo Fisher Scientific) at 20 V for 7 min. The PVDF membranes were then blocked with 5% non-fat skim milk. The protein blots were incubated with anti-ARID1A antibody (dilution, 1:1000; catalog no., #12354; Cell Signaling Technology, Danvers, MA, USA), anti-EZH2 antibody (dilution, 1:1000; catalog no., #5246; Cell Signaling Technology), anti-phosphorylated (p)-AKT Ser473 (dilution, 1:2000; #4060; Cell Signaling Technology), anti-AKT (dilution, 1:2000; catalog no., #4691; Cell Signaling Technology), anti-pPI3K (dilution, 1:1000; catalog no., #4228; Cell Signaling Technology), and anti-PI3K (dilution, 1:1000; catalog no., #4257; Cell Signaling Technology), overnight at 4 °C, followed by incubation with goat anti-rabbit HRP secondary antibody (Santa Cruz Biotechnology, Dallas, Texas, USA) for 1 h at room temperature. The protein levels were quantified using primary mouse anti-β-actin antibody (dilution, 1:2000; catalog no., #sc-69789; Santa Cruz Biotechnology) as the internal loading control for 1 h at room temperature. Bound antibodies were detected using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific), visualized by autoradiography (ImageQuant™ LAS 4000 IR MultiColor imager; Fujifilm Corporation, Tokyo, Japan) and quantified with Image J software (Version 1.52s, National Institutes of Health, Bethesda, MD, USA).
TCGA database analysis
Gene mutation or expression data of GC cell lines or GC patients were obtained from the Catalogue of Somatic Mutations in Cancer (COSMIC) cell line project (https://cancer.sanger.a.uk) or TCGA’s cBioPortal database (https://www.cbioportal.org/). Those data of ARID1A in GC cells and of ARID1A, EZH2, PIK3IP1, and PI3K/AKT signaling genes in GC patients were used for the analyses. For the expression data, RNA-sequencing data that was normalized by RSEM method was used for the analyses. For the clinical tumors, a multi-omics study including 293 GC patients was selected for the analyses [3]. Among 293 GC tumors, 198 tumors were ARID1A-wildtype (WT) and 95 tumors were ARID1A-mutations (MUT), including 78 tumors with ARID1A truncating mutations. Furthermore, 183 out of 198 tumors with ARID1A-WT and 70 out of 78 tumors with ARID1A truncating mutations were used in this study due to the full data availability. For PI3K/AKT signaling genes, expression data of PI3K subunit phosphoinositide-3-kinase, regulatory subunit 1 (PIK3R1) and subunit 2 (PIK3R2) and phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA), AKT isoform AKT1 and AKT3, mammalian target of rapamycin (mTOR), and forkhead box O (FOXO) subtype FOXO3 and FOXO4 were used for the analyses.
Patient samples
The study included 39 surgical specimens from ARID1A-deficient patients from our previous study [5]. Because ARID1A mutations are frequently detected in GC patients with microsatellite instability (MSI)-high/DNA mismatch repair deficiency (dMMR) and Epstein–Barr virus (EBV)-positive patients (who often show PD-L1 and PD-L2 overexpression), we analyzed the expression of MMR, EBV, PD-L1, and PD-L2 in ARID1A-deficient GC patients. Data on age, sex, TNM stage (8th classification), and pathological diagnosis, including lymphatic and venous invasion, were retrospectively collected. The carcinomas at the time of primary tumor resection were staged according to the Union for International Cancer Control classification. The study was approved by the ethics committee of Fukushima Medical University. All patients provided written informed consent.
Immunohistochemical staining and evaluation
Immunohistochemical (IHC) staining for ARID1A, EZH2, and PD-L1 was performed in our previous study [5] and PD-L2 staining was additionally performed in this study. IHC staining of paraffin-embedded histological sections (4 μm thick) was performed using a polymer peroxidase method. Briefly, after deparaffinization and rehydration, the sections were treated with 0.3% hydrogen peroxide in methanol for 30 min to block endogenous peroxidase activity. After rinsing in PBS, the sections were incubated with anti-ARID1A antibody (#12354; D2A8U; 1:500 dilution; Cell Signaling Technology), anti-EZH2 antibody (#5246; D2C9; 1:50; Cell Signaling Technology), anti-PD-L1 antibody (#13684; E1L3N; 1:400; Cell Signaling Technology), and anti-PD-L2 antibody (#82723; D7U8C; 1:200; Cell Signaling Technology) at 4 °C overnight. An additional wash in PBS was followed by treatment with a peroxidase-labeled polymer conjugated to goat anti-rabbit immunoglobulins (ENvision + kit; Dako, Agilent, Santa Clara, CA, USA) as the secondary antibody for 30 min at room temperature. The staining was visualized with diaminobenzidine, followed by counterstaining with hematoxylin. Protein expression was considered positive when the nucleus of the cancerous tissue and the total field of view were observed at 400 × magnification. Loss of ARID1A protein was defined as the absence of nuclear staining in tumor cells in the presence of positive nuclear staining in stroma cells. Weak or partial positive staining for ARID1A was not defined as positive staining. Specimen staining was evaluated by investigators blinded to the origin of samples and clinical outcomes. Stained cancer cells were counted per 1000 cancer cells in the maximum tumor tissue field by two investigators. Positive staining rates were classified as follows: 0%, 0; 1–10%, 1; and 11–100%, 2. The staining intensity was scored as 0 (negative), 1 (weak), and 2 (strong). The results were expressed as a product of the positivity score and staining intensity score. Positive staining was defined by a score of 2, whereas negative staining (ARID1A-deficient) was defined by a score of 0 or 1. Evaluation of PD-L1/L2 expression was performed as previously described [24]. Positive expression of the PD-L1/L2 proteins was defined as positive staining in GC cells.
Determination of MMR status
IHC staining for MMR proteins was performed for all the cases in the test cohort as previously described [5], and the following primary antibodies were used: MLH1 (ES05, 1:50, Dako), MSH2 (FE11, 1:50, Dako), MSH6 (EP49, 1:200, Dako), and PMS2 (EP51, 1:50, Dako). Loss of an MMR protein was defined as absence of nuclear staining in tumor cells in the presence of positive nuclear staining in normal epithelial cells and lymphocytes. Tumors showing loss of at least one MMR protein were collectively designated as dMMR, and tumors with intact MMR protein expression were designated as pMMR.
EBV encoding region (EBER) in situ hybridization (ISH) and evaluation
Integration of EBV was assessed by ISH as previously described [5], and ISH was completed for all cases in this study. EBER-ISH was performed using the INFORM EBER probe (800–2842, Ventana Medical Systems, Tucson, AZ, USA) and the Discovery XT autostainer (Ventana) according to the manufacturer’s protocol.
Statistical analysis
Mann–Whitney U test were used to determine differences between two variables. Spearman’s correlation was used to evaluate the correlations between levels of expression. All statistical analyses were conducted using GraphPad Prism v7.0 (Graphpad Software Inc., San Diego, CA, USA). All P values were two-sided, and P values less than 0.05 were considered statistically significant.
Results
ARID1A-MUT GC cells are sensitive to EZH2 inhibitors
Based on the synthetic lethal approach, we investigated the selectivity of EZH2 inhibitors for ARID1A-deficient GC cells. To measure the sensitivity of cells to EZH2 inhibitors, four GC cell lines [two ARID1A-wild-type (ARID1A-WT) and two ARID1A-mutated (ARID1A-MUT) cell lines] were selected and confirmed ARID1A mutation profiles from COSMIC cell line project database. First, in addition to measuring the expression of ARID1A, we confirmed the protein expression of EZH2 in each GC cell line because the response to EZH2 inhibitors often correlates with EZH2 overexpression [20]. Consistent with COSMIC database, ARID1A expression was positive in MKN7 and NCI-N87 (ARID1A-WT) cells, whereas it was negative in OCUM-1 (ARID1A-MUT) cells (Fig. 1a). Although ARID1A expression was positively detected in NUGC-3 (ARID1A-MUT) cells, it seemed that ARID1A mutations in NUGC-3 cells are heterozygous mutant, leads to the production of non-functional protein.

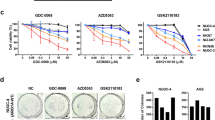
Cell viability of ARID1A-wildtype (WT) and ARID1A-mutated (MUT) gastric cancer (GC) cell lines against EZH2 inhibitors. a Western blot analysis of ARID1A and EZH2 expressions in whole cell extracts of four GC cell lines. MKN7 and NCI-N87 (ARID1A-WT) cells showed positive ARID1A expression and OCUM-1 (ARID1A-MUT) cells showed negative ARID1A expression. Although NUGC-3 (ARID1A-MUT) cells showed positive ARID1A expression, it seemed that ARID1A mutations leads to the production of non-functional protein. EZH2 expression was detected in all cell lines. β-actin was used as a loading control. Cell viability of ARID1A-WT MKN7 and ARID1A-MUT NUGC-3 cell lines (b) and ARID1A-WT NCI-N87 and ARID1A-MUT OCUM-1 cell lines (c) by cell proliferation assays. The concentrations of EZH2 inhibitors used were as follows: GSK126: 0.04, 0.2, 1, and 5 μM: GSK343: 0.04, 0.2, 1, and 5 μM: and EPZ005687: 0.31, 0.93, 2.8, and 8.3 μM. Each value represents the mean ± SD of three independent experiments. NS, not significant. *P < 0.05, **P < 0.001, and ***P < 0.0001. d Comparison of the cell viability by colony formation assays of MKN7, NCI-N87 and NUGC-3 cells including negative control (NC) and treatment for 14 days with 5 μM GSK126, 5 μM GSK343, and 8.3 μM EPZ005687. Cells were not colonized in EZH2 inhibitors treated NUGC-3 cells. e Quantification of the number of colonies is expressed in all field of each well in d
To evaluate the selective sensitivity of ARID1A-deficient GC cells to EZH2 inhibitors, the viability and survival of ARID1A-WT MKN7 and ARID1A-MUT NUGC-3 cells, that were adhesion cell type, was measured. The three EZH2 inhibitors, GSK126, GSK343, and EPZ005687, are inhibitors of PRC2, whereas they show differential selectivity for EZH2 [16]. Although treatment with three EZH2 inhibitors decreased cell viability in both cell lines, the effect was markedly stronger in NUGC-3 cells than in MKN7 cells (Fig. 1b). To further confirm the selective sensitivity of ARID1A-deficient GC cells to EZH2 inhibitors, the viability and survival were measured in ARID1A-WT NCI-N87, that was adhesion cell type, and ARID1A-MUT OCUM-1 cells, that was floating cell type. Consistent with the results in Fig. 1b, although treatment with three EZH2 inhibitors decreased cell viability in a weak dose-dependent manner in both cell lines, the effect was markedly stronger in OCUM-1 cells than in NCI-N87 cells (Fig. 1c). Colony formation assays were performed to measure cell survival in adhesion cell types of MKN7, NCI-N87, and NUGC-3 cells, which demonstrated the selective sensitivity of NUGC-3 cells to EZH2 inhibitors (Fig. 1d). Quantification of the colony formation assays showed that EZH2 inhibitors significantly suppressed colony growth in NUGC-3 cells, while the finding was not observed in MKN7 and NCI-N87 cells (Fig. 1e). These results suggest that ARID1A-MUT GC cells are selectively sensitive to EZH2 inhibitors.
Selective sensitivity to EZH2 inhibitors in ARID1A knockdown GC cells
To further determine whether the selective sensitivity of cells to EZH2 inhibitors was related to ARID1A deficiency, cell viability was measured after transient ARID1A knockdown in ARID1A-WT GC cells. First, we confirmed that two different siRNA oligonucleotides against ARID1A efficiently downregulated ARID1A expression in ARID1A-WT MKN7 and ARID1A-WT NCI-N87 cells, and that EZH2 expression was preserved in ARID1A knockdown cells (Fig. 2a, b), in line with the previous study [5].

Cell viability of ARID1A-WT MKN7 and NCI-N87 cells in which ARID1A was silenced by EZH2 inhibitors. a ARID1A and EZH2 expressions in ARID1A-WT (MKN7 and NCI-N87) cells in which ARID1A was silenced using siRNAs. Western blot analysis of ARID1A and EZH2 expressions in MKN7 and NCI-N87 cells by ARID1A silencing (siARID1A #1 and #2 and NC, negative control). β-Actin was used as a loading control. b Quantitative results of ARID1A and EZH2 bands by ImageJ software. Each protein expression level was normalized to β-actin values. Both siRNAs mediated significant decrease of ARID1A and preserved EZH2 expressions in MKN7 and NCI-N87 cells. Comparison of the viability of ARID1A knockdown cells and control cells by cell proliferation assays in MKN7 (c) and NCI-N87 (d) cells. Absorbance results at 450 nm (OD450nm) in the whole cell assays and quantification of cell viability results. The concentrations of EZH2 inhibitors were as follows: GSK126: 0, 0.04, 0.2, 1, and 5 μM: GSK343: 0, 0.04, 0.2, 1, and 5 μM: and EPZ005687: 0, 0.31, 0.93, 2.8, 8.3 μM. ARID1A knockdown cells showed a dose-dependent decrease of cell viability and lower cell viability than the negative control. Each value represents the mean ± SD of three independent experiments. Relative cell viability of ARID1A knockdown cells and control cells in each concentration of EZH2 inhibitors in MKN7 (e) and NCI-N87 (f) cells. The concentrations of EZH2 inhibitors were as follows: GSK126: 0.04, 0.2, 1, and 5 μM: GSK343: 0.04, 0.2, 1, and 5 μM: and EPZ005687: 0.31, 0.93, 2.8, 8.3 μM. Three EZH2 inhibitors showed markedly stronger effect with dose-dependent manner in ARID1A knockdown cells. Each value represents the mean ± SD of three independent experiments. *P < 0.05, **P < 0.001, and ***P < 0.0001
Next, we investigated the effect of EZH2 inhibitors on the viability of ARID1A knockdown MKN7 and NCI-N87 cells. The results showed that the three EZH2 inhibitors decreased the viability of cells with transient ARID1A knockdown compared with that of control cells in the MKN7 (Fig. 2c) and NCI-N87 (Fig. 2d) cell lines. The treatment effect with three EZH2 inhibitors was markedly stronger in a nearly dose-dependent manner in the MKN7 (Fig. 2e) and NCI-N87 (Fig. 2f) cell lines. These results confirmed the selective sensitivity of ARID1A-deficient GC cells to EZH2 inhibitors, which suggested that EZH2 synthetic lethal therapy could be developed as a therapeutic strategy for ARID1A-deficient GC.
PI3K/AKT signaling is a candidate for an actionable target of EZH2 inhibitor in ARID1A-deficient GC
It was reported that ARID1A and EZH2 are commonly targetable for PIK3IP1 and regulate the PI3K/AKT signaling pathway [12]. PIK3CA, an important regulator of the PI3K/AKT pathway, is frequently co-mutated with ARID1A and cooperates with ARID1A to promote tumor growth [25]. In ARID1A-proficient cells, we hypothesized that the homeostasis between ARID1A and EZH2 can maintain the PI3K/AKT signaling pathway (Fig. 3a). On the other hand, in ARID1A-deficient cells, due to the homeostatic mechanisms between ARID1A and EZH2 are disrupted, the PI3K/AKT signaling pathway is supposed to be activated via PIK3IP1, possibly leading to enhancement of tumor growth (Fig. 3b).

Schematic representation of proposed mechanism behind ARID1A, EZH2 and PI3K/AKT signaling pathway. a In ARID1A-proficient cells, PIK3IP1 can suppress PI3K/AKT signaling showing normal cell growth. b In ARID1A-deficient cells, PIK3IP1 is supposed to be downregulated by EZH2, leading to the activation of PI3K/AKT signaling and upregulation of cell proliferation and survival. c EZH inhibition by EZH2 inhibitor in ARID1A-deficient cells can restore the expression of PIK3IP1, resulting in downregulation of PI3K/AKT signaling. EZH2 inhibitor can induce antitumor effect in ARID1A-deficient cells. PI3K, phosphatidylinositol-3-kinase; PIK3IP1, phosphoinositide-3-kinase interacting protein 1; AKT, protein kinase B; mTOR, mammalian target of rapamycin; FOXO, forkhead box O
In order to evaluate this hypothesis in ARID1A-proficient and -deficient GC, we accessed TCGA’s cBioPortal database and investigated the expression of PI3K/AKT signaling genes. First, we confirmed that 41 of 293 (14.0%, P < 0.001) GC patients in this cohort harbored both ARID1A and PIK3CA mutations in the line with a previous report [25]. Then, the expression data of PI3K subunits, PIK3R1, PIK3R2 and PIK3CA, AKT isoforms, AKT1 and AKT3, mammalian target of rapamycin (mTOR), and FOXO subtypes regulating apoptosis, FOXO3 and FOXO4, were compared between ARID1A-WT (n = 183) and ARID1A-MUT (n = 70) (limited to tumors with ARID1A truncating mutations) GC tumors (Supplementary Table S1). As a result, the PIK3R2 and AKT1 expressions were significantly upregulated in ARID1A-MUT GCs in comparison to those in ARID1A-WT GCs, while FOXO3 that induce apoptosis, were downregulated in ARID1A-MUT GCs, suggesting the possibility of the PI3K/AKT activation by ARID1A deficiency as like in our hypothesis in Fig. 3a, b, (Fig. 4a). To further determine whether ARID1A deficiency effect on the activation of PI3K/AKT signaling pathway, pPI3K and pAKT were measured by western blotting after transient ARID1A knockdown in ARID1A-WT GC cells. In the line with our hypothesis, pAKT was slightly upregulated in ARID1A-deficient cells than control cell (Fig. 4b). These results supported that the PI3K/AKT signaling was tended to be activated in ARID1A-MUT GC in comparison to ARID1A-WT GC.

Evaluation of activation and expression in PI3K/AKT signaling genes stratified by ARID1A mutation status or EZH2 expressions. a The mRNA expression of PI3K/AKT signaling genes, PIK3IP1, PI3K (PIK3R1, PIK3R2, and PIK3CA), AKT (AKT1 and AKT3), mTOR, and FOXO (FOXO3 and FOXO4), were compared between GC patients with ARID1A-wildtype (WT) (n = 183) and ARID1A truncating mutation (MUT) (n = 70). PIK3R2 and AKT1 expressions were significantly increased in GC patients with ARID1A-MUT compared to those with ARID1A-WT. Expression of AKT3 and FOXO3 were significantly decreased in GC patients with ARID1A-MUT compared to those with ARID1A-WT. b Western blot analysis of phosphorylated (p)AKT (Ser473), AKT, pPI3K, and PI3K expressions in ARID1A-WT (MKN7 and NCI-N87) cells in which ARID1A was silenced using siRNAs (siARID1A #1 and #2 and NC, negative control). β-Actin was used as a loading control. c Correlation of EZH2 mRNA expression with PI3K/AKT signaling genes in GC patients with ARID1A-MUT (n = 70). Expression between EZH2 and PIK3IP1 was inversely correlated. Expression of PIK3R2 and mTOR were positively and AKT3 and FOXO4 was negatively correlated with EZH2 expression. Data were provided by The Cancer Genome Atlas (Project ID; TCGA-STAD). NS, not significant. *P < 0.05, **P < 0.001, and ***P < 0.0001
Next, in order to mimic the possible effect of EZH2 inhibitors in ARID1A-MUT GC as shown in Fig. 3c, correlation of EZH2 mRNA expression with the PI3K/AKT signaling genes in ARID1A-MUT GC tumors (n = 70) was evaluated. As a result, significant negative correlation was found between EZH2 and PIK3IP1 mRNA expressions (Fig. 4c). Furthermore, PIK3R2 and mTOR expressions were positively correlated with EZH2 expression. Also, FOXO4 expression was negatively correlated with EZH2 expression. These results indirectly suggested that down-regulation of EZH2 in GC patients with ARID1A-MUT is related to downregulation of PI3K/AKT signaling pathway.
Discussion
The present study demonstrated that the synthetic lethal approach using EZH2 inhibitors could be a useful therapeutic strategy for the treatment of ARID1A-deficient GC. This novel strategy is based on the findings that selective sensitivity of EZH2 inhibition using three kinds of specific EZH2 inhibitors for ARID1A-deficient GC cells (ARID1A-MUT and ARID1A-WT GC cells that transiently knocked down of ARID1A) was firmly confirmed.
EZH2 inhibition is promising therapeutic efficacy against ARID1A-deficient cancer that are dependent on a non-catalytic role of EZH2, suggesting partially dependent on EZH2 histone methyltransferase activity [20]. It is suggested that, in ARID1A-proficient cells, both ARID1A and EZH2 directly target PIK3IP1, which suppresses PI3K/AKT signaling, keeping homeostasis cell growth. On the other hand, in the ARID1A-deficient cancer, it is likely that EZH2 suppresses PIK3IP1, leading to the activation of PI3K/AKT signaling and enhancing cell viability. In the present study, the gene expression data obtained from TCGA database also suggested the PI3K/AKT signaling was activated in ARID1A-deficient GC in comparison to ARID1A-proficient GC. We therefore performed in vitro cell experiments and confirmed the activation of PI3K/AKT signaling that pAKT was induced by ARID1A deficiency. Furthermore, as a postulated mechanism underlying selective sensitivity of EZH2 inhibitors in ARID1A-deficient cancer, TCGA data showed that decreased expression of EZH2 correlated with the suppression of PI3K/AKT signaling in ARID1A-deficient GC, that are mimicry corresponding to EZH2 inhibition against ARID1A-deficient GC. In other words, downregulation of EZH2 in ARID1A-deficient GC is possibly related to downregulation of PI3K/AKT signaling and control of tumor cell growth.
Furthermore, an additional therapeutic target of histone deacetylase 2 (HDAC2), interact with EZH2-containing PRC2 complex, has been reported in ARID1A-mutated ovarian cancer [26]. Since HDAC2 works as a co-repressor of EZH2 in ARID1A-deficient cells, HDAC inhibitors can inhibit cell proliferation and promote apoptosis via restoration of PIK3IP1 in ARID1A-deficient cells. These results again support the hypothesis that the homeostasis between EZH2 and ARID1A is related to PI3K/AKT signaling pathway, and EZH2 inhibition in ARID1A-deficient cells can control tumor cell growth. Further study regarding the molecular analysis in EZH2 inhibitor-treated GC cells with ARID1A-deficiency will be needed.
Of interest, EZH2 inhibitors also decreased cell viability in ARID1A-proficient (ARID1A-WT in Fig. 1b, c or control cells in Fig. 2c, d) GC cells, although the effect was markedly prominent in ARID1A-deficient cells. Originally, it is known that the enzymatic inhibition of EZH2 methyltransferase activity is considered to show a therapeutic efficacy for only cancers with EZH2 overexpression or gain of function mutations [17,18,19]. In fact, some of clinical trials (e.g., NCT01897571 and NCT03155620) with EZH2 inhibitors, including GSK126, have been started for patients with various cancers, but not limited for those of ARID1A-deficient cancers [16]. It is likely that there is other actionable mechanism of EZH2 inhibitors, although EZH2 inhibition based on synthetic lethality in ARID1A-deficient GC is a reasonable theory.
To further examine the clinical relevance of our results, we examined the clinicopathological characteristics of ARID1A-deficient GC patients (Supplementary Fig. S1 and Supplementary Table S2) [5]. The results suggested that EZH2 inhibitors could be beneficial in most ARID1A-deficient GC patients (36 of 39 cases, 92.3%), whereas three cases showed negative EZH2 expression and would not benefit from EZH2 inhibitors. To determine whether immune checkpoint inhibitors could be beneficial for ARID1A-deficient GC patients, especially for EZH2 negative patients, MSI-high/dMMR, EBV-positivity, and PD-L1/L2 overexpression were evaluated based on prior studies [27, 28]. Most of the ARID1A-deficient GCs showed positive expression of PD-L1 and/or PD-L2 and two cases without EZH2 expression in ARID1A-deficent GCs also showed PD-L1 and PD-L2 expression. These observations suggested that ARID1A-defient GC patients might be treated with immune checkpoint inhibitors in addition to EZH2 inhibitors. In fact, recent studies reported that ARID1A directly represses CD274 gene transcription, and loss of ARID1A triggers PD-L1 upregulation through the PI3K/AKT signaling pathway in GC cells [29, 30].
Next generation sequencing-based panel sequencing is currently used in daily clinical practice, which could improve the detection of ARID1A mutations in various cancers as well as in GC [31]. In this study, since ARID1A-deficient GC cells are shown to be sensitive to EZH2 inhibitors, a synthetic lethality approach with EZH2 inhibitors should be considered as an additional therapeutic strategy for ARID1A-dificient GC.
References
Jim MA, Pinheiro PS, Carreira H, Espey DK, Wiggins CL, Weir HK. Stomach cancer survival in the United States by race and stage (2001–2009): findings from the CONCORD-2 study. Cancer. 2017;123(Suppl 24):4994–5013.
Taniyama Y, Katanoda K, Charvat H, Hori M, Ohno Y, Sasazuki S, et al. Estimation of lifetime cumulative incidence and mortality risk of gastric cancer. Jpn J Clin Oncol. 2017;47:1097–102.
Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9.
Selim JH, Shaheen S, Sheu WC, Hsueh CT. Targeted and novel therapy in advanced gastric cancer. Exp Hematol Oncol. 2019;8:25.
Ashizawa M, Saito M, Min AKT, Ujiie D, Saito K, Sato T, et al. Prognostic role of ARID1A negative expression in gastric cancer. Sci Rep. 2019;9:6769.
Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–23.
Wilson MR, Reske JJ, Holladay J, Wilber GE, Rhodes M, Koeman J, et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat Commun. 2019;10:3554.
Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3:35–433.
Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. 2017;355:1152–8.
Ogiwara H, Takahashi K, Sasaki M, Kuroda T, Yoshida H, Watanabe R, et al. Targeting the vulnerability of glutathione metabolism in ARID1A-deficient cancers. Cancer Cell. 2019;35(177–90):e8.
Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med. 2014;20:251–4.
Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. 2015;21:231–8.
Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015;5:752–67.
Williamson CT, Miller R, Pemberton HN, Jones SE, Campbell J, Konde A, et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun. 2016;7:13837.
Bitler BG, Wu S, Park PH, Hai Y, Aird KM, Wang Y, et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat Cell Biol. 2017;19:962–73.
Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34.
Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–6.
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–12.
Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA. 2012;109:21360–5.
Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med. 2015;21:1491–6.
Ujiie D, Okayama H, Saito K, Ashizawa M, Thar Min AK, Endo E, et al. KRT17 as a prognostic biomarker for stage II colorectal cancer. Carcinogenesis. 2019. https://doi.org/10.1093/carcin/bgz192.
Yu T, Wang Y, Hu Q, Wu W, Wu Y, Wei W, et al. The EZH2 inhibitor GSK343 suppresses cancer stem-like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget. 2017;8:98348–59.
Geissmann Q. OpenCFU, a new free and open-source software to count cell colonies and other circular objects. PLoS ONE. 2013;8:e54072.
George J, Saito M, Tsuta K, Iwakawa R, Shiraishi K, Scheel AH, et al. Genomic amplification of CD274 (PD-L1) in small-cell lung cancer. Clin Cancer Res. 2017;23:1220–6.
Abe H, Kaneda A, Fukayama M. Epstein–Barr virus-associated gastric carcinoma: use of host cell machineries and somatic gene mutations. Pathobiology. 2015;82:212–23.
Fukumoto T, Park PH, Wu S, Fatkhutdinov N, Karakashev S, Nacarelli T, et al. Repurposing pan-HDAC inhibitors for ARID1A-mutated ovarian cancer. Cell Rep. 2018;22:3393–400.
Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. 2018;24:1449–588.
Sasaki S, Nishikawa J, Sakai K, Iizasa H, Yoshiyama H, Yanagihara M, et al. EBV-associated gastric cancer evades T-cell immunity by PD-1/PD-L1 interactions. Gastric Cancer. 2019;22:486–96.
Kim YB, Ahn JM, Bae WJ, Sung CO, Lee D. Functional loss of ARID1A is tightly associated with high PD-L1 expression in gastric cancer. Int J Cancer. 2019;145:916–26.
Fukumoto T, Fatkhutdinov N, Zundell JA, Tcyganov EN, Nacarelli T, Karakashev S, et al. HDAC6 inhibition synergizes with anti-PD-L1 therapy in ARID1A-inactivated ovarian cancer. Cancer Res. 2019;79:5482–9.
Saito M, Momma T, Kono K. Targeted therapy according to next generation sequencing-based panel sequencing. Fukushima J Med Sci. 2018;64:9–14.
Acknowledgements
The work was supported by JSPS KAKENHI Grant numbers 18K08652 and 19K09099.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yamada, L., Saito, M., Thar Min, A.K. et al. Selective sensitivity of EZH2 inhibitors based on synthetic lethality in ARID1A-deficient gastric cancer. Gastric Cancer 24, 60–71 (2021). https://doi.org/10.1007/s10120-020-01094-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-020-01094-0