
Abstract
Autosomal recessive congenital ichthyosis (ARCI) is a group of keratinisation disorders that includes the ichthyosis prematurity syndrome (IPS). IPS is rare and almost exclusively present in a restricted region in the middle of Norway and Sweden, which indicates a founder effect for the disorder. We recently reported linkage of IPS to chromosome 9q34, and we present here the subsequent fine-mapping of this region with known and novel microsatellite markers as well as single nucleotide polymorphisms (SNPs). Allelic association, evaluated with Fisher’s exact test and P excess, was used to refine the IPS haplotype to approximately 1.6 Mb. On the basis of the average length of the haplotype in IPS patients, we calculated the age of a founder mutation to approximately 1,900 years. The IPS haplotype contains a core region of 76 kb consisting of four marker alleles shared by 97.7% of the chromosomes associated with IPS. This region spans four known genes, all of which are expressed in mature epidermal cells. We present the results from the analysis of these four genes and their corresponding transcripts in normal and patient-derived samples.
Similar content being viewed by others

Introduction
Autosomal recessive congenital ichthyosis (ARCI) is a clinically and genetically heterogeneous group of disorders that affect the keratinisation process of the skin (Williams and Elias 1985). ARCI can be subdivided into at least four groups (I–IV) by electron microscopy (Anton-Lamprecht 1992). So far, eight loci have been associated with ARCI (chromosomes 2q34, 3p21, 5q33, 9q33-34, 14q11, 17p13, 19p12-q12 and 19p13), and a corresponding disease gene has been identified in six of these regions (Russell et al. 1994; Parmentier et al. 1996; Fischer et al. 2000; Virolainen et al. 2000; Krebsova et al. 2001; Jobard et al. 2002; Klar et al. 2004; Lefevre et al. 2004, 2006).
Ichthyosis prematurity syndrome (IPS), also known as ichthyosis congenita type IV (Anton-Lamprecht 1992), is a distinct form of ARCI. IPS is characterised by premature birth, usually at gestational weeks 30–32, of the affected child, which has a thick desquamating epidermis. Asphyxia is a frequent complication, probably due to aspiration of amnion debris. Soon after the critical neonatal period the child’s health improves spontaneously. No developmental abnormalities are present, and the skin symptoms become minor, but there are a slightly thickened epidermis and follicular hyperkeratosis, which remain into adulthood. IPS is very rare, except in a region in Norway and Sweden, with an estimated heterozygote carrier frequency of 1 in 50 (Gedde-Dahl et al. 1999). This indicates an ancient founder mutation for IPS in the area. IPS has been reported in three families outside Norway and Sweden, of which two are from Finland and one from Italy (Niemi et al. 1993; Brusasco et al. 1997). We recently reported linkage of IPS to chromosome 9q33.3–34.13 (Klar et al. 2004), and a shared ancestral haplotype was expected among a majority of the affected individuals because of the geographical clustering. In this study we present the refinement of the IPS locus, using a highly dense marker haplotype. From the results we calculated the age of an ancient founder mutation and analysed specific candidate genes within a 76 kb core region (Hastbacka et al. 1994; Engert et al. 2000; Joensuu et al. 2001).
Materials and methods
Participants
Blood samples were collected from 22 families with at least one member affected with IPS. Siblings were available for 19 out of the 22 families, of which 13 had been previously reported with linkage to chromosome 9q34 (Klar et al. 2004). The six new families were added to expand the previous linkage study, and all 22 families were included in the haplotype analysis. Altogether, 28 affected and 22 healthy siblings were available for analysis. A majority of the families originated from a defined region in middle Norway and Sweden (Fig. 1). None of the families was known to be related, and no consanguinity could be ascertained. Affected families were clinically examined (Norwegian families by T.G.-D., Swedish families by A.V. and a Danish family by F.B. and A.B.), and ultrastructure of skin biopsies was determined by electron microscopy. All affected family members presented with a phenotype consistent with IPS, including premature birth. Informed consent was obtained from all participants, and the study was approved by the ethics committee at Uppsala University.

Map of Norway and Sweden. The shaded portion shows the geographical area from which the majority of the IPS families originated. IPS has the highest prevalence in the city of Trondheim
Genotyping
DNA was extracted from whole blood, and genotyping was performed using fluorescence-labelled microsatellite markers. Genotype data for 15 markers at the IPS locus were available from the previous linkage study, and an additional 22 polymorphic repeats, of which 12 are novel, were used as markers in the linked interval between marker loci D9S250 and D9S63 (Klar et al. 2004). We amplified the markers by polymerase chain reaction (PCR) and incorporated fluorescent labels, as previously described (Klar et al. 2004). Genotypes for four single nucleotide polymorphisms (SNPs) were obtained through sequencing, as described elsewhere (Entesarian et al. 2005). Altogether, 41 marker loci, spaced at a mean distance of 150 kb, were included in the analysis. Additional SNPs were genotyped in families IR71 and SV76 in order to refine the minimal shared haplotype. Primer sequences and allele frequencies for the novel markers are available upon request.
Linkage and haplotype analysis
Two point LOD score calculations were made using the MLINK program of the LINKAGE software package (version 5.1) (Lathrop and Lalouel 1984). We assumed an autosomal recessive inheritance, an equal male and female recombination rate and full penetrance. The disease allele frequency was set to 1 per 150,000 due to a proportion of obligate carriers born in geographical areas with a reduced incidence of IPS.
Haplotypes were drawn using Cyrillic (version 2.1.3) (Cherwell Scientific Publishing, Oxford, UK) and “affected” and “unaffected” control haplotypes were constructed for the association studies using sizes of marker alleles in affected children and their parents for genetically linked markers. The aggregation of families in a distinct region suggests a high local carrier frequency, and unaffected parental haplotypes were used as controls. This corresponds to the transmission disequilibrium test. Associations between allele sizes and the disease phenotype were calculated with Fisher’s exact test and P excess (Hastbacka et al. 1994).
The age of a major founder mutation for IPS was estimated from the mean length of the disease haplotype identified in affected individuals. This is calculated as 2/g morgans, where g is the number of generations after the mutation was funded (Boehnke 1994). This equation has previously been consistent with empirical data (Engert et al. 2000) and can be used for estimating the age of the mutation.
Mutation and expression analysis
Sequencing analysis of all exons and flanking intron regions of TBC1D13, ENDOG, C9Orf114, CCBL1 located within the critical interval was performed on three affected individuals (IR61, IR125 and SV76). Expressed sequence tags (ESTs) and mRNA sequences were utilised as a means to identify new putative exons for the previously known transcripts within the region as well as a way to predict new, independent, transcribed sequences. This was done by aligning all human ESTs and mRNA sequences to the genomic assembly (NCBI, build 35.1) using BLAT (Kent 2002). The genomic alignments were converted into GFF files and manually inspected in the Apollo browser (Lewis et al. 2002) for identification of novel expressed transcripts.
The ECR browser was used to identify additional candidate regions of possible functional importance from the degree of conservation between human, dog, mouse and rat (Ovcharenko et al. 2004). Regions with a conservation level >65% over 100 bp were selected. Conserved sequences and regions with homology to expressed ESTs were sequenced as described previously (Entesarian et al. 2005).
Primary keratinocytes from three IPS patients homozygous for the haplotype (IR104, IR122 and IR125, Fig. 3) and three healthy controls were cultured in a DMEM/F12 medium, with a feeder layer of radiated 3T3 cells. The cells were maintained at 37°C in a humidified atmosphere with 5% CO2. RNA was prepared using TRIzol (Invitrogen, San Diego, Calif., USA) according to the manufacturer’s protocol. The quality of the total RNA was controlled by capillary electrophoresis using a Bioanalyzer 2100 (Agilent Technologies, Palo Alto, Calif., USA). Only RNA with no sign of degradation was used. cDNA was prepared using 2 μg total RNA, 6 μl MMLV buffer (5×), 0.3 μl BSA (1 μg/μl), 0.25 μl HPRI (31 U/μl), 2 μl dNTP (10 mM), 1 μl random primers (25 μm) and 0.45 μl MMLV (200 U/μl) in a 30 μl reaction. The reaction mix was heated to 50°C without MMLV, cooled to room temperature, at which point MMLV was added, incubated at 37°C for 1 h and finally incubated at 80°C for 10 min.
Oligonucleotide primers positioned within the transcripts for TBC1 domain family member 13 (TBC1D13), endonuclease G (ENDOG), chromosome 9 open reading frame 114 (C9Orf114) and cysteine conjugate-beta lyase (CCBL1) were designed (supplementary Fig. 1). Exon splicing was investigated with PCR, spanning all known exon boundaries specific for each of the four transcripts (supplementary Fig. 1) using cDNA from one patient (IR122) and two controls. The Platinum SYBR Green qPCR SuperMix-UDG kit was used for quantitative real time PCR according to the manufacturer’s protocol (Invitrogen). cDNA levels of β-actin (ACTB) and GAPDH were used for normalisation. The PCR and detection were performed with the ABI PRISM 7000 Sequence Detection System (Applied Biosystems). All oligonucleotide primers were designed with the Primer3 program (http://www.frodo.wi.mit.edu/). PCR conditions used are available upon request.
Results
Linkage analysis
Linkage analysis confirmed previous linkage of IPS to chromosome 9q33.3–34.13 (Klar et al. 2004). A maximum cumulative LOD score of 5.3 was obtained with marker D9S752 (θ=0) for 19 families. The centromeric recombination event is defined by marker D9S282, which further restricts the IPS locus in this study to a 5.8 Mb interval (approximately 6 cM) between markers D9S282 and D9S63.
Association and haplotype studies
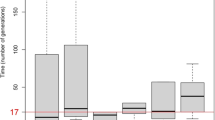
The region between the closest flanking recombinations was further investigated with a total number of 37 microsatellite markers, which were analysed with Fisher’s exact test in order to assess association of markers to the disease. This resulted in significant P values in two different regions, spanning 1.1 Mb and 2.0 Mb, respectively. Within these regions allelic excess was calculated for each marker locus used as a method to identify a haplotype associated with the disease (Fig. 2). Three adjacent microsatellite markers (CAA13-ATTT10-D9S260) showed strong allelic excess (P excess≥0.9), which defined a possible core haplotype (marker alleles 2-2-1, respectively) in the region (Fig. 3). Three SNP markers (rs6478853, rs6478854 and rs11542344) confirmed this core haplotype. This six-marker haplotype was present on 91% of the disease-associated chromosomes and on 23% of the control chromosomes (Table 1). The distribution resulted in an allelic excess of 0.88. The six markers constituted the core in a common haplotype block (Table 1), spanning, on average, 2.56 cM (1.60 Mb) in the affected individuals (Fig. 3). Based on this average haplotype length in affected individuals, the age of the mutation in our population was calculated to 78 generations. With a mean generation age of 25 years (Genin et al. 2004), the IPS mutation was introduced in the population approximately 1,900 years ago.

Association results for microsatellite markers in the region restricted by recombination events. The Y-axis denotes 1-p for Fisher’s exact test and P excess for allelic excess. The X-axis denotes the physical position in Mb (NCBI, build 35.1). Black bars annotate regions showing allelic association spanning 1.1 Mb and 2.0 Mb, respectively. Allelic excess peaks are achieved for alleles of markers CAA13, ATTT10 and D9S260

Haplotypes from a 1.8 Mb region on chromosome 9q34 in 22 independent IPS patients. A haplotype (shaded) can be constructed in the affected individuals around the markers rs6478853, rs6478854, CAA13, ATTT10, rs11542344 and D9S260. Positions, in megabases, are from the sequence tagged site map of GenBank (NCBI, build 35.1). Paternal haplotype is the first column of each individual and maternal haplotype in the second
Three patients, IR71, SV76 and IR61, are heterozygous for the six-marker haplotype (Fig. 3). Patients IR71 and SV76 are homozygous for markers rs6478853, rs6478854, CAA13 and ATTT10, which suggests that the IPS mutation is restricted to this region. IR71 and SV76 are both heterozygous for rs10988141, which restricts the minimal shared haplotype towards the telomere (Fig. 3). The shared region is restricted towards the centromere by D9S61 in nine patients. The paternal haplotype of IR61 shares only one single SNP marker allele with the associated haplotype (Fig. 3). The minimal ancestral haplotype in all affected patients but IR61 restricts the IPS candidate region to a maximum 76 kb. To date, four genes are known to be located in this region: TBC1D13, ENDOG, C9Orf114 and CCBL1.
Mutation and expression analysis
Genomic sequencing of DNA from three patients, IR61, IR125 and SV76, revealed no sequence alterations in the exons of the four genes TBC1D13, ENDOG, C9Orf114 and CCBL1. Twenty base pairs of intronic sequences flanking each exon were found to be normal when compared to public databases. Four evolutionary conserved regions (ECRs), ranging from 100 bp to 271 bp were extracted from the ECR database (Table 2) (Ovcharenko et al. 2004). Three new possible 5′UTRs were identified upstream of CCBL1 from the EST data, one of which coincided with an ECR. In addition, a region of approximately 700 bp in intron 1 of ENDOG was supported by several ESTs as an independent transcript (Table 2). These seven regions were all sequenced but showed no variations from sequences in the public databases.
In search of aberrant splicing due to mutations in introns we performed PCR on cDNA using primers spanning all known exon boundaries in each of the four transcripts. The PCR products showed similar sizes when samples from patient and controls were compared (data not shown). The four genes are expressed in a wide variety of tissues, including keratinocytes, as verified by quantitative PCR on cDNA from cultured keratinocytes. The analysis revealed no significant differences in expression levels between patients and controls (Fig. 4). Student’s t-test for TBC1D13, ENDOG, C9Orf114 and CCBL1 yielded P values of 0.12, 0.48, 0.88 and 0.34, respectively.

Quantitative real-time PCR. Normalised mRNA levels in cultured keratinocytes are shown for patients (grey,n=3) and controls (black, n=3) on a logarithmic scale. No significant differences in expression level between patients and controls were detected
Discussion
In this report we map the IPS by allelic and haplotype association to a 1.6 Mb region on chromosome 9q34. Within this interval we identified a core region, defined by six marker alleles, forming a haplotype shared by 91% of the IPS chromosomes analysed. This indicates a strong founder effect for IPS, and the age of the founder mutation is calculated to approximately 1,900 years. Three patients show deviating haplotypes in the core region. Two of these patients are homozygous for four of the six markers, which suggests that the core haplotype is restricted to this interval. The third patient carries one deviating haplotype with one single marker allele shared by the core haplotype for IPS. There are several possible explanations for this: the deviating haplotype may be very small and not detected at this marker resolution. Other possibilities are: mutations in marker loci, resulting in altered allele variants, or that the patient is compound heterozygous for two IPS mutations.
The size of a shared chromosomal region allowed us to calculate the age of the IPS founder mutation. The size of the shared segment is predominantly determined by the total number of meioses to a common ancestor. With the use of microsatellite markers it is important to consider their mutation rate, which is in the range of 10−4 to 10−2 per locus and generation (Lai and Sun 2003; Ellegren 2004). In this example we estimated that an ancestral mutant chromosome has passed through 78 generations, and it is likely that mutations have occurred at some of the marker loci, which reduces their absolute frequency among the affected individuals. This may be one reason for IPS-associated haplotypes interrupted by single microsatellite markers (IR85 and IR107, Fig. 3). On the other hand, this observation could also be the result of recombinations between similar or identical haplotypes. The calculations of haplotype length are affected by the mutation rate of polymorphic alleles, the marker density and the background allele frequencies. As a consequence, this influences the estimated age of the IPS mutation in this population. From our results, the average length of the haplotype identified corresponds to a founder mutation with an age of approximately 1,900 years.
The core haplotype restricts a region of 76 kb containing four genes (TBC1D13, ENDOG, C9Orf114, CCBL1) all shown to be expressed in keratinocytes. Endonuclease G is involved in DNA cleavage at a stage in mitochondrial DNA replication. It also has a function in chromosomal degradation during apoptosis (Cote and Ruiz-Carrillo 1993; Li et al. 2001). The cysteine conjugate-beta lyase is also known as kynurenine aminotransferase I and glutamine transaminase K, due to its functions in different reaction pathways (Cooper and Meister 1974; Okuno et al. 1996). Little is known about the functions of the TBC1D13 and C9Orf114 gene products. A majority of the genes found to be mutated in ARCI encodes proteins involved in lipid transportation or modification, such as the 12(R)-lipoxygenase pathway (Jobard et al. 2002; Lefevre et al. 2006). No connections could be found between the TBC1D13, ENDOG, CCBL1 or C9Orf114 gene products and pathways previously shown to be involved in ARCI. We found no sequence alterations in the exons of the four genes, or in evolutionary conserved or EST supported regions within the 76 kb. Significant differences in transcript levels were excluded between patients and controls for TBC1D13, ENDOG, CCBL1 or C9Orf114, indicating a normal expression of the genes in the IPS patients (Fig. 4). In addition, we found no indications of aberrant splicing by the analysis of intron sequences flanking each exon of the four genes or by cDNA PCR spanning all exon boundaries in each of the four transcripts (data not shown).
Possible explanations for a yet unidentified IPS mutation may be a larger rearrangement, such as an inversion, insertion or duplication which mediates a positional effect. The mutation may also affect a regulatory region, controlling a gene outside the 76 kb. If conserved, a regulatory region could be smaller than the ECR analysed in this study. A third possibility is that the mutation is located within a region encoding a small non-coding RNA. Their functions include regulation of transcription, RNA processing and translation and stability of mRNAs (Storz 2002). Small RNAs, such as microRNAs and snoRNAs, are generally well-conserved across species, but since they are short (typically 21–25 nt and 60–300 nt, respectively) they may have escaped detection in our ERC and EST analysis (Pang et al. 2006). The 76 kb core region will be further investigated in search of structural changes and short functional motifs.
In this study we define the IPS mutation to a 76 kb region containing four candidate genes. Haplotype analysis confirms a strong founder effect for the disorder. Refinement of the IPS haplotype has facilitated prenatal diagnostics, and carrier screening in families with affected members and diagnosis will be further improved by the identification of the IPS gene mutation. Moreover, this may bring clues to mechanisms in normal skin formation and factors initiating child delivery.
References
Anton-Lamprecht I (1992) The skin. In: Papadimitrou J, Henderson D, Spagnolo D (eds) Diagnostic ultrastructure of non-neoplastic diseases. Churchill and Livingstone, Edinburgh, pp 459–550
Boehnke M (1994) Limits of resolution of genetic linkage studies: implications for the positional cloning of human disease genes. Am J Hum Genet 55:379–390
Brusasco A, Gelmetti C, Tadini G, Caputo R (1997) Ichthyosis congenita type IV: a new case resembling diffuse cutaneous mastocytosis. Br J Dermatol 136:377–379
Cooper AJ, Meister A (1974) Isolation and properties of a new glutamine transaminase from rat kidney. J Biol Chem 249:2554–2561
Cote J, Ruiz-Carrillo A (1993) Primers for mitochondrial DNA replication generated by endonuclease G. Science 261:765–769
Ellegren H (2004) Microsatellites: simple sequences with complex evolution. Nat Rev Genet 5:435–445
Engert JC, Berube P, Mercier J, Dore C, Lepage P, Ge B, Bouchard JP, Mathieu J, Melancon SB, Schalling M, Lander ES, Morgan K, Hudson TJ, Richter A (2000) ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet 24:120–125
Entesarian M, Matsson H, Klar J, Bergendal B, Olson L, Arakaki R, Hayashi Y, Ohuchi H, Falahat B, Bolstad AI, Jonsson R, Wahren-Herlenius M, Dahl N (2005) Mutations in the gene encoding fibroblast growth factor 10 are associated with aplasia of lacrimal and salivary glands. Nat Genet 37:125–127
Fischer J, Faure A, Bouadjar B, Blanchet-Bardon C, Karaduman A, Thomas I, Emre S, Cure S, Ozguc M, Weissenbach J, Prud’homme JF (2000) Two new loci for autosomal recessive ichthyosis on chromosomes 3p21 and 19p12-q12 and evidence for further genetic heterogeneity. Am J Hum Genet 66:904–913
Gedde-Dahl T Jr, Pigg M, Dahl N (1999) Prehistoric mutation causes the ichthyosis–prematurity syndrome (ichthyosis congenita type IV) in Middle Norway. Abstract presented at the European Science Foundation meeting, Obernai
Genin E, Tullio-Pelet A, Begeot F, Lyonnet S, Abel L (2004) Estimating the age of rare disease mutations: the example of Triple-A syndrome. J Med Genet 41:445–449
Hastbacka J, de la Chapelle A, Mahtani MM, Clines G, Reeve-Daly MP, Daly M, Hamilton BA, Kusumi K, Trivedi B, Weaver A, et al (1994) The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell 78:1073–1087
Jobard F, Lefevre C, Karaduman A, Blanchet-Bardon C, Emre S, Weissenbach J, Ozguc M, Lathrop M, Prud’homme JF, Fischer J (2002) Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum Mol Genet 11:107–113
Joensuu T, Hamalainen R, Yuan B, Johnson C, Tegelberg S, Gasparini P, Zelante L, Pirvola U, Pakarinen L, Lehesjoki AE, de la Chapelle A, Sankila EM (2001) Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet 69:673–684
Kent WJ (2002) BLAT—the BLAST-like alignment tool. Genome Res 12:656–664
Klar J, Gedde-Dahl T Jr, Larsson M, Pigg M, Carlsson B, Tentler D, Vahlquist A, Dahl N (2004) Assignment of the locus for ichthyosis prematurity syndrome to chromosome 9q33.3–34.13. J Med Genet 41:208–212
Krebsova A, Kuster W, Lestringant GG, Schulze B, Hinz B, Frossard PM, Reis A, Hennies HC (2001) Identification, by homozygosity mapping, of a novel locus for autosomal recessive congenital ichthyosis on chromosome 17p, and evidence for further genetic heterogeneity. Am J Hum Genet 69:216–222
Lai Y, Sun F (2003) The relationship between microsatellite slippage mutation rate and the number of repeat units. Mol Biol Evol 20:2123–2131
Lathrop GM, Lalouel JM (1984) Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet 36:460–465
Lefevre C, Bouadjar B, Karaduman A, Jobard F, Saker S, Ozguc M, Lathrop M, Prud’homme JF, Fischer J (2004) Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Hum Mol Genet 13:2473–2482
Lefevre C, Bouadjar B, Ferrand V, Tadini G, Megarbane A, Lathrop M, Prud’homme JF, Fischer J (2006) Mutations in a new cytochrome P450 gene in lamellar ichthyosis type 3. Hum Mol Genet 15:767–776
Lewis SE, Searle SM, Harris N, Gibson M, Lyer V, Richter J, Wiel C, Bayraktaroglir L, Birney E, Crosby MA, Kaminker JS, Matthews BB, Prochnik SE, Smithy CD, Tupy JL, Rubin GM, Misra S, Mungall CJ, Clamp ME (2002) Apollo: a sequence annotation editor. Genome Biol 3:Research0082
Li LY, Luo X, Wang X (2001) Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412:95–99
Niemi KM, Kuokkanen K, Kanerva L, Ignatius J (1993) Recessive ichthyosis congenita type IV. Am J Dermatopathol 15:224–228
Okuno E, Nishikawa T, Nakamura M (1996) Kynurenine aminotransferases in the rat. Localization and characterization. Adv Exp Med Biol 398:455–464
Ovcharenko I, Nobrega MA, Loots GG, Stubbs L (2004) ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res 32:W280–W286
Pang KC, Frith MC, Mattick JS (2006) Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet 22:1–5
Parmentier L, Lakhdar H, Blanchet-Bardon C, Marchand S, Dubertret L, Weissenbach J (1996) Mapping of a second locus for lamellar ichthyosis to chromosome 2q33–35. Hum Mol Genet 5:555–559
Russell LJ, DiGiovanna JJ, Hashem N, Compton JG, Bale SJ (1994) Linkage of autosomal recessive lamellar ichthyosis to chromosome 14q. Am J Hum Genet 55:1146–1152
Storz G (2002) An expanding universe of noncoding RNAs. Science 296:1260–1263
Virolainen E, Wessman M, Hovatta I, Niemi KM, Ignatius J, Kere J, Peltonen L, Palotie A (2000) Assignment of a novel locus for autosomal recessive congenital ichthyosis to chromosome 19p13.1-p13.2. Am J Hum Genet 66:1132–1137
Williams ML, Elias PM (1985) Heterogeneity in autosomal recessive ichthyosis. Clinical and biochemical differentiation of lamellar ichthyosis and nonbullous congenital ichthyosiform erythroderma. Arch Dermatol 121:477–488
Acknowledgements
In memory of T.G.-D., who died in February 2006 during the preparation of this manuscript. The authors thank the patients and their families for their contribution to this study. We acknowledge Marja Boström, Department of Surgical Sciences at Uppsala University Hospital for assistance in cell culturing. This work was supported by grants from the Swedish Research Council, Children’s Cancer Foundation of Sweden, Swedish Cancer Society, Swedish Medical Society, Torsten and Ragnar Söderbergs Fund, Borgström Foundation, Welander-Finsen Foundation, Network for Ichthyoses and Keratinization Disorders of the German Ministry of Education and Research. The collection of the Norwegian families over two decades was supported initially by Nansenfondet og de dermed forbundne fonds, University of Oslo, and later by the Anders Jahres Fund’s general support to basic research at the Institute of Forensic Medicine, University of Oslo.
Author information
Authors and Affiliations
Corresponding author
Additional information
T. Jr Gedde-Dahl: deceased in February 2006.
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Melin, M., Klar, J., Jr Gedde-Dahl , T. et al. A founder mutation for ichthyosis prematurity syndrome restricted to 76 kb by haplotype association. J Hum Genet 51, 864–871 (2006). https://doi.org/10.1007/s10038-006-0035-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0035-z
Keywords
This article is cited by
-
Inherited ichthyoses/generalized Mendelian disorders of cornification
European Journal of Human Genetics (2013)
-
Ichthyin/NIPAL4 localizes to keratins and desmosomes in epidermis and Ichthyin mutations affect epidermal lipid metabolism
Archives of Dermatological Research (2012)
-
Development of an Ichthyosiform Phenotype in Alox12b-Deficient Mouse Skin Transplants
Journal of Investigative Dermatology (2009)
