
Abstract
ATP-sensitive potassium (KATP) channels, composed of pore-forming Kir6.2 and regulatory sulphonylurea receptor (SUR) subunits, play an essential role in insulin secretion from pancreatic beta cells. Binding of ATP to Kir6.2 inhibits, whereas interaction of Mg-nucleotides with SUR, activates the channel. Heterozygous activating mutations in Kir6.2 (KCNJ11) are a common cause of neonatal diabetes (ND). We assessed the functional effects of six novel Kir6.2 mutations associated with ND: H46Y, N48D, E227K, E229K, E292G, and V252A. KATP channels were expressed in Xenopus oocytes and the heterozygous state was simulated by coexpression of wild-type and mutant Kir6.2 with SUR1 (the beta cell type of SUR). All mutations reduced the sensitivity of the KATP channel to inhibition by MgATP, and enhanced whole-cell KATP currents. Two mutations (E227K, E229K) also enhanced the intrinsic open probability of the channel, thereby indirectly reducing the channel ATP sensitivity. The other four mutations lie close to the predicted ATP-binding site and thus may affect ATP binding. In pancreatic beta cells, an increase in the KATP current is expected to reduce insulin secretion and thereby cause diabetes. None of the mutations substantially affected the sensitivity of the channel to inhibition by the sulphonylurea tolbutamide, suggesting patients carrying these mutations may respond to these drugs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
This issue of Pflugers Archiv celebrates the 25th anniversary of a milestone in electrophysiology—the development of giga-seal patch clamp recording [1]. This technique has not only confirmed that ion channels are the molecular entities responsible for the electrical activity of excitable cells; it has elucidated, in detail, how ion channels work [2]. Equally importantly, it has increased our understanding of the role of ion channels in physiological functions of cells as diverse as neurones, lymphocytes, and pancreatic beta cells [2, 3] and revealed that mutations in ion channel genes underlie a wide spectrum of human diseases [3, 4]. About 20 years ago, the patch clamp technique was used to demonstrate that ATP-sensitive potassium (KATP) channels play a key role in insulin secretion from pancreatic beta cells [5]. In this paper, we use patch clamp methods to show how mutations in the KATP channel give rise to impaired insulin secretion and neonatal diabetes.
Neonatal diabetes is usually diagnosed within the first 6 months of life [6]. In some patients, the diabetes is transient, although it may frequently relapse, whereas in other cases it is permanent and requires insulin treatment for life. The commonest cause of permanent neonatal diabetes (PNDM) are heterozygous activating mutations in the KCNJ11, the gene encoding Kir6.2, which constitutes the pore-forming subunit of the KATP channel in the pancreatic beta cell [7–9]. KCNJ11 mutations account for around 50% of cases of PNDM [7–13]. Some of these mutations lead to a more severe syndrome in which developmental delay and epilepsy accompany neonatal diabetes, a condition known as DEND syndrome [9]. Mutations in KCNJ11 can also result in transient neonatal diabetes mellitus (TNDM) [14], although in most patients TNDM is caused by an abnormality of the imprinted region on chromosome 6q24 [15]. Kir6.2 associates with the sulphonylurea receptor SUR1 (ABCC8) to form the KATP channel of the pancreatic beta cell [16, 17] and mutations in ABCC8 can also cause neonatal diabetes [18].
The beta cell KATP channel is a hetero-octameric complex of Kir6.2 and SUR1 [19]. Four Kir6.2 subunits form a central tetrameric pore, and they are surrounded by four regulatory SUR1 subunits [20], which modulate the opening and closing (gating) of the pore. The most important modulators of KATP channel gating are adenine nucleotides: in particular, ATP closes the channel by binding to Kir6.2, whereas interaction of Mg-nucleotides (MgATP, MgADP) with the nucleotide-binding domains (NBDs) of SUR1 stimulates channel activity and reverses channel inhibition by ATP [21–23]. Nucleotide modulation enables KATP channels to couple changes in plasma glucose levels to changes in insulin secretion [24, 25].
At sub-stimulatory glucose concentrations, K+ efflux through open KATP channels holds the beta cell membrane at a hyperpolarized potential, so preventing the opening of voltage-gated Ca2+ channels, calcium influx, and insulin secretion (Fig. 1). Increased plasma glucose stimulates glucose uptake and metabolism by the beta cell and leads to an increase in ATP and an accompanying decrease in MgADP that results in KATP channel closure. This produces insulin release by promoting membrane depolarization, opening of voltage-gated Ca2+ channels, and Ca2+ influx [24, 25].

Cartoon illustrating how the KATP channel couples glucose metabolism to insulin secretion [24]. a When metabolism is low, KATP channels are open, so that the beta cell is hyperpolarized and electrical activity and insulin secretion are prevented. b Glucose enters the cell and via glycolytic and mitochondrial metabolism leads to an increase in ATP and fall in MgADP in the immediate vicinity of the KATP channel. This results in KATP channel closure, membrane depolarization, opening of voltage-gated Ca2+ channels, Ca2+ influx, and exocytosis of insulin granules. c If the ATP sensitivity of the KATP channel is impaired, ATP elevation fails to close the channel, so that the membrane remains hyperpolarized and insulin secretion is prevented
Mutations in KATP channel subunits that reduce channel function lead to congenital hyperinsulinism, whereas those that enhance channel function cause diabetes [8, 9]. To date, mutations in Kir6.2 associated with TNDM and PNDM have been shown to enhance KATP channel activity by decreasing ATP inhibition at Kir6.2 [7–9, 13, 14, 26–31]. In addition, they may also increase channel activation by Mg-nucleotides, by enhancing the stimulatory action of SUR1 on Kir6.2 [28, 29].
KATP channels are also the target for sulphonylurea drugs, which are widely used to treat type 2 diabetes [32]. These drugs stimulate insulin secretion by binding to SUR1, and closing KATP channels directly, thus bypassing beta cell metabolism. They have proved effective in treating neonatal diabetes that results from gain-of-function mutations in Kir6.2 [9, 10, 33].
In this paper, we examine the functional effects of six novel missense mutations recently reported to be associated with neonatal diabetes in the Spanish population: H46Y, N48D, E227K, E229K, V252A, and E292G. These mutations are associated with TNDM (N48D, E227K, E229K), PNDM (H46Y, E292G) or both (V252A).
Materials and methods
Human Kir6.2 (Genbank NM000525 with E23 and I337) and rat SUR1 (Genbank L40624) were used in this study. Site-directed mutagenesis of Kir6.2, preparation of mRNA, and isolation of Xenopus laevis oocytes was performed as described previously [29, 34]. Oocytes were injected with 0.8 ng wild-type or mutant Kir6.2 mRNA and ∼4 ng of SUR1 mRNA (giving a 1:5 ratio). To simulate the heterozygous state, SUR1 was coexpressed with a 1:1 mixture of wild type and mutant Kir6.2 (28). Currents were recorded 1–3 days after injection.
Whole-cell currents were recorded using the two-electrode voltage-clamp method in response to voltage steps of ±20 mV from a holding potential of −10 mV [34]. They were filtered at 1 kHz and digitized at 4 kHz. Oocytes were constantly perfused at 20–22°C with a solution containing (in mM): 90 KCl, 1 MgCl2, 1.8 CaCl2, and 5 HEPES (pH 7.4 with KOH). Metabolic inhibition was produced by 3 mM Na-azide and the sulphonylurea tolbutamide (0.5 mM) was used to confirm currents flowed through KATP channels.
Macroscopic currents were recorded from giant excised inside-out patches using the patch-clamp technique in response to 3-s voltage ramps from −110 to +100 mV (holding potential, 0 mV) at 20–22°C. Currents were filtered at 0.15 kHz and digitised at 0.5 kHz. The pipette solution contained (mM): 140 KCl, 1.2 MgCl2, 2.6 CaCl2, 10 HEPES (pH 7.4 with KOH). The internal (bath) solution contained (mM): 107 KCl, 1 CaCl2, 2 MgCl2, 10 EGTA, 10 HEPES (pH 7.2 with KOH) and MgATP as indicated. For single-channel recordings, we used the same external solution and the internal solution contained (in mM): 107 KCl, 1 K2SO4, 10 EGTA, 10 HEPES (pH 7.2 with KOH).
The macroscopic slope conductance was measured between −100 and +10 mV. ATP concentration–response curves were fit with the Hill equation:
where [ATP] is the ATP concentration, IC50 is the ATP concentration at which inhibition is half maximal and h is the slope factor (Hill coefficient). To account for possible rundown, Gc was taken as the mean of the conductance in control solution before and after ATP application. Concentration–response curves were fitted individually for each patch and the mean±SEM of the individual parameters are given in Table 1. The curves shown in the figures are the best fit of Eq. 1 to the mean data points.
Single channel currents were recorded at −60 mV, filtered at 2 kHz, and digitized at 20 kHz. Open probability was determined as previously described [26, 27].
All data were analyzed with pCLAMP8 (Axon Instruments, CA, USA), Origin 6.02 (Microcal Software, Northampton, MA, USA) and Igor (Wavemetrics, Lake Oswego, OR, USA) software, and are given as mean±SEM. Statistical significance was evaluated using an unpaired two-tailed Student t test. A probability value of P<0.05 was taken as the criteria for a significant difference.
Results
Location of mutations
Figure 2a illustrates the position of the mutated residues in a structural model of Kir6.2 [35]. In the 3-D structural model (Fig. 2b), H46 and N48 lie on a flexible loop that connects the cytosolic N terminus of the channel to the slide helix, an amphipathic helix that lies in the plane of the membrane and is postulated to be involved in gating of the channel [36]. Both residues lie in close proximity to residue R50, which is predicted to interact with the γ-phosphate of ATP (Fig. 2c). In agreement with this prediction, mutations at R50 impair ATP inhibition [30, 37–39] and can give rise to neonatal diabetes [12, 30]. Mutation of either H46 or N48 might, therefore, be expected to influence the position of R50, and thereby destabilize ATP binding. Residue V252 lies within the C-terminal domain of the channel, in a region that comes into close proximity with the N terminus. Interestingly, this residue lies within 4.0Å of H46 (Fig. 2b). Thus mutation of V252 and H46 might influence KATP channel ATP sensitivity by a similar mechanism.

Location of mutations in Kir6.2. a Molecular model of Kir6.2 [35], viewed from the side. For clarity, each subunit is shown in a different colour and only two transmembrane domains (TMs) and two cytosolic domains are illustrated. Mutated residues are shown in ball-and-stick format and ATP in yellow. b Close-ups of the interactions made by H46, N48, and V252. c Close-up of the ATP-binding site showing the positions of E292 and three residues that interact with ATP (R50, R201, and R301). d Close-ups of the interactions made by E227 and E229 (orange) with residues in the adjacent subunit (mustard). In b–d, residues mutated in neonatal diabetes are shown in red and ATP in yellow
Residue E292 is predicted to form an inter-subunit ion pair with R301, which makes a backbone interaction with the adenine ring of ATP (Fig. 2c). E292 also lies within one of the two gating loops that link the ATP-binding site to the slide helix, and which may, therefore, be involved in coupling ATP binding to channel opening [35, 40]. Thus, disruption of the putative E292-R301 ion pair can be hypothesized to either alter the mechanism by which ATP binding is linked to channel gating, or to destabilize ATP binding indirectly, by altering the conformation of the ATP-binding site.
Finally, the model predicts that E227 and E229 in one Kir6.2 subunit form putative ion pairs with residues in the adjacent subunit (R192 and R314, respectively). Thus, E227 and E229 may be involved in electrostatic interactions between subunits (Fig. 2d). Their mutation to a positively charged residue would be expected to break these interactions and thereby might influence channel gating. Indeed, functional studies have previously shown that E229 makes an ion pair with R314 that is critical for maintaining channel activity [41].
Effects on whole-cell currents
Because all patients are heterozygotes, their pancreatic beta cells will contain a mixture of wild-type and mutant Kir6.2. We, therefore, explored the functional effects of Kir6.2 mutations on KATP channel function in the simulated heterozygous state, by coinjecting a 1:1 mixture of mutant and wild-type Kir6.2 together with SUR1 [28]. This will produce a mixed population of homomeric wild-type channels, homomeric mutant channels and heteromeric channels containing between 1 and 3 mutant subunits. We refer to this channel population as heterozygous channels.
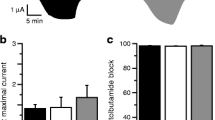
When expressed in Xenopus oocytes, wild-type Kir6.2/SUR1 channels are normally closed due to the high intracellular ATP concentration ([ATP]i), and open only in response to metabolic inhibitors, such as azide, which lower [ATP]i (Fig. 3a,b; [34]). Heterozygous channels usually produced resting currents that were slightly greater than wild-type, further increased by azide, and substantially blocked by the sulphonylurea tolbutamide (Fig. 3a,c). In most cases, the increase in resting current, relative to wild type, was very small. The percentage block of the azide-activated current by tolbutamide varied from 82 to 96%, being not substantially different from that found for wild-type channels (96%; Fig. 3c).

Comparison of whole-cell currents of wild-type and mutant KATP channels. a Whole-cell currents recorded from Xenopus oocytes coexpressing SUR1 and either Kir6.2 or both Kir6.2 and Kir6.2-E227K (hetE227K), in response to voltage steps of ±20 mV from a holding potential of −10 mV. The bars indicate the times of application of 3 mM azide and 0.5 mM tolbutamide (tolb). b Mean steady-state whole-cell currents evoked from oocytes coexpressing SUR1 plus wild-type (WT) and mutant Kir6.2, as indicated, by a voltage step from −10 to −30 mV. To control for small differences in expression between different batches of oocytes, currents are expressed as a percentage of their amplitude in the presence of 3 mM azide. The number of oocytes is given below the bars. c Percentage KATP current inhibition by 0.5 mM tolbutamide for wild-type (WT) and mutant KATP channels (as indicated), measured in the presence of 3 mM azide. The number of oocytes is given below the bars
Effects on KATP channel ATP sensitivity
We next explored the effect of the mutations on the sensitivity of heterozygous channels to inhibition by MgATP. Experiments were carried out in the presence of 2 mM Mg2+ to most closely approximate the physiological condition.
Figure 4 shows that all mutant heterozygous channels had impaired ATP sensitivity. The concentration of ATP causing half maximal block (IC50) shifted from 15 μM for wild-type channels to 41 μM for V252A channels and as much as 213 μM for E227K channels (Table 1). There was also a marked increase in the current amplitude in the presence of 1 mM MgATP, which increased between tenfold for E229K and 30-fold for N48D (Table 1). Likewise, a large increase in current was observed in the presence of 3 mM MgATP (Table 1).

ATP sensitivity of wild-type and mutant channels a KATP currents recorded in response to successive voltage ramps from −110 to +100 mV in an inside-out patch excised from a Xenopus oocyte expressing Kir6.2/SUR1 (WT) and heterozygous Kir6.2-H46Y/SUR1 KATP channels. The dashed line indicates the zero current level. The bar indicates application of 1 mM ATP. b Mean relationship between [ATP] and KATP conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc) for Kir6.2/SUR1 (○, n=6), and heterozygous mutant Kir6.2/SUR1 channels (●), as indicated. The smooth curves are the best fit of Eq. 1 to the data. The number of patches for mutant channels is given in Table 1
In the case of the E227K and E229K mutations, the heterozygous currents ran down rapidly after patch excision into nucleotide-free solution. However, they increased substantially after application of high concentrations of MgATP (>1 mM), as previously reported for homomeric channels with mutations at E229 [41]. The ATP sensitivity was measured following activation with high MgATP concentrations.
Effects of the mutations on the intrinsic open probability
Previous studies have shown that mutations in Kir6.2 can reduce ATP inhibition of the KATP channel by several mechanisms. For example, they may disrupt ATP binding or impair the mechanism by which ATP binding is translated into closure of the channel pore [26, 29, 30]. They may also affect ATP sensitivity indirectly by stabilizing the intrinsic (unliganded) open state of the KATP channel [26, 27]. This shifts the gating equilibrium in the presence of ATP towards the open state, and, therefore, reduces its inhibitory effect [42, 43].
To determine whether the Kir6.2 mutations that lie outside the putative ATP binding site mutation affect the intrinsic open probability [Po(0)], we recorded single-channel currents from wild-type and mutant homomeric channels from inside-out membrane patches in nucleotide-free solution (Fig. 5a). Because homomeric currents ran down quickly in patches excised into Mg2+ and nucleotide-free solution, Po(0) was measured during the first 10 s after excision. The Po(0) of E227K and E229K channels was significantly greater (p<0.05) than that of wild-type channels, being 0.72±0.02 (n=5) for E227K and 0.68±0.04 (n=5) for E229K, compared with 0.38±0.05 (n=10) for the wild-type channel. There was no effect on the single-channel conductance. The H46Y mutation, which lies within the ATP-binding site, had no significant effect on Po(0), which was 0.32±0.04 (n=8).

a Single-channel currents recorded at −60 mV from inside-out membrane patches excised from oocytes expressing Kir6.2/SUR1, Kir6.2-H46Y/SUR1, and Kir6.2-E227K/SUR1. The dashed lines indicate the zero current level. b Decline of channel activity observed for Kir6.2/SUR1 Kir6.2-E229K/SUR1 following patch excision. The patch was excised just before the recording. The dashed lines indicate the zero current level
Despite the increased Po(0), the homomeric E227K and E229K currents ran down faster than wild type (Fig. 5b). The rate of rundown could be well fit with a single exponential with a time constant of 41±10 s (n=5) for E227K and 29±9 s (n=5) for E229K, compared with 135±67 s (n=6) for wild-type channels. These values are approximately five- to tenfold slower than those reported for mutation of E229 to lysine, alanine or cysteine [41].
Discussion
Our results demonstrate that the six Kir6.2 mutations associated with PNDM or TNDM that we have studied are gain-of-function mutations, which act by reducing the ability of MgATP to inhibit the KATP channel. This adds to accumulating evidence that KCNJ11 mutations that cause neonatal diabetes do so by affecting KATP channel ATP sensitivity, either directly or indirectly [8, 9, 13, 14, 26–30].
Four of the six mutations studied, H46Y, N48D, V252A, and E292G, are found close to the predicted ATP-binding site. Thus, it seems likely that mutation of these residues impairs ATP binding, probably by allosterically altering the conformation of the binding site itself. The lack of effect of the H46Y mutation on the single-channel kinetics is consistent with this idea. Two mutations, E227K and E229K, lie distant from the ATP-binding site at the interface between adjacent subunits. These mutations alter the channel ATP sensitivity indirectly by increasing the intrinsic open probability of the channel [42, 43]. The increase in Po(0) (to 0.68 and 0.72) is similar to that found for mutations at residue F35, which cause PNDM (∼0.75, (31)), but less than that observed for mutations associated with DEND syndrome (∼0.85, [26, 27]).
The data presented in this paper further emphasize that it is the magnitude of the increase in the KATP current in the presence of physiologically relevant concentrations of ATP that determines the severity of the clinical phenotype, and not the mechanism of action of the mutation. Thus, mutations that impair the gating of the channel can cause TNDM, as shown here, as well as PNDM [31] and DEND syndrome [26, 27]. When Po(0) is greater than about ∼0.8, very small changes in P(0) can have large effects on ATP sensitivity because of the steep relationship between Po(0) and the IC50 for ATP inhibition [43]. Mutations that lie in the ATP-binding site can also produce different clinical phenotypes, depending on the extent to which they impair the channel ATP sensitivity [30].
Effects of reduced ATP sensitivity
The reduction in ATP sensitivity produced by the KCNJ11 mutations we studied is associated with a small increase in the resting KATP current when expressed in Xenopus oocytes. Although in some cases the increase in resting current was not significant, this may not necessarily be the case in pancreatic beta cells, where resting ATP concentrations appear to be lower and native wild-type KATP channels are open in the absence of glucose. An increase in KATP current may be expected to hyperpolarize the beta cell membrane and reduce or abolish the membrane depolarization evoked by glucose. This will prevent electrical activity, calcium influx and insulin secretion, and could thereby account for the diabetic phenotype of the patients.
Our results suggest neonatal diabetes was produced by rather small changes in the ability of MgATP to block the KATP channel: for example, a 2.7-fold reduction in the IC50 (V252A) and a fivefold increase in the current at 1 mM MgATP (E229K). This is consistent with previous reports that diabetes can be associated with small changes in nucleotide sensitivity in man [14, 26, 29–31] and mouse [44]. Given that the beta cell membrane potential is largely determined by the activity of the KATP channel, it is not surprising that small changes in ATP sensitivity, which produce tiny changes in KATP current, can result in marked changes in electrical activity, and insulin secretion [25].
Genotype-phenotype correlations
We found no clear correlation between the clinical phenotype (TNDM and PNDM) and the magnitude of the reduction in ATP sensitivity of heterozygous KATP channel, whether ATP sensitivity was measured as the IC50 or as the current amplitude in the presence of 3 mM MgATP (Table 1). A comparison of all neonatal diabetes mutations studied to date (Fig. 6) reveals that this is also the case for other mutations associated with TNDM and PNDM. Furthermore, the V252A mutation was associated with TNDM in one patient and with PNDM in others, highlighting the importance of the genetic background of the different patients and/or the role of environmental influences [34]. Differences in clinical phenotype associated with the same mutations have also been reported previously [8, 9].

Fractional current remaining in the presence of 3 mM MgATP, measured in inside-out patches for wild-type (WT) KATP channels and KATP channels containing the indicated Kir6.2 mutations. Pale grey bars indicate mutations associated with TNDM, dark gray bars mutations causing PNDM, and black bars mutations producing DEND syndrome. The number of patches varied from 6 to 9. Data are taken from this paper and published work [26, 27, 29–31]
It is important to note, however, that with a single exception (N48D), the unblocked currents in the presence of 3 mM MgATP (7–10%) lay within the range of values previously reported for TNDM and PNDM mutations. Furthermore, they were much smaller than those found for Kir6.2 mutations that give rise to a DEND syndrome, where the unblocked current was between 28 and 40% (Fig. 6).
Clinical implications
The reason why some mutations cause a relapsing remitting form of diabetes whereas others cause permanent diabetes is unclear. As previously suggested, this may be due to a reduced insulin requirement at the time of remission; to changes in beta cell turnover due to the Kir6.2 mutation; or to compensatory changes (at the level of the beta cell, pancreas, or whole body), which are able to overcome the effects of the channel defect.
The ability of tolbutamide to block the whole-cell currents in oocytes ranged from 82 to 96%, compared with 96% for wild-type channels (Fig. 3c). This suggests that it should be possible to control the diabetes of all the patients described here with sulphonylureas, as has been successfully achieved for Kir6.2 mutations, which cause a similar degree of reduction in ATP sensitivity [7, 9, 10, 33, 45].
References
Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch 391:85–100
Hille B (2001) Ion channels of excitable membranes, 3rd Ed. Sinauer Associates. 814 pages
Ashcroft FM (2000) Ion channels and disease. Academic. 481 pages
Ashcroft FM (2006) Ion channels and disease: from molecule to malady. Nature 440:440–447
Ashcroft FM, Harrison DE, Ashcroft SJH (1984) Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature 312:446–448
Shield JP (2000) Neonatal diabetes: new insights into aetiology and implications. Horm Res 53(Suppl 1):7–11
Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining J, Slingerland AS, Howard N, Srinivasan S, Silva JMCL, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JPH, Sumnik Z, van Rhijn A, Wales JKH, Clark P, Gorman S, Aisenberg J, Ellard S, Njølstad PR, Ashcroft FM, Hattersley A (2004) Activating mutations in the ATP-sensitive potassium channel subunit Kir6.2 gene are associated with permanent neonatal diabetes. New Eng J Med 350:1838–1849
Ashcroft FM (2005) ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest 115:2047–2058
Hattersley AT, Ashcroft FM (2005) Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights and new therapy. Diabetes 54:2503–2513
Sagen JV, Raeder H, Hathout E, Shehadeh N, Gudmundsson K, Baevre H, Abuelo D, Phornphutkul C, Molnes J, Bell GI, Gloyn AL, Hattersley AT, Molven A, Sovik O, Njolstad PR (2004) Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes 53:2713–2718
Vaxillaire M, Populaire C, Busiah K, Cave H, Gloyn AL, Hattersley AT, Czernichow P, Froguel P, Polak M (2004) Kir6.2 mutations are a common cause of permanent neonatal diabetes in a large cohort of French patients. Diabetes 53:2719–2722
Massa O, Iafusco D, D’Amato E, Gloyn AL, Hattersley AT, Pasquino B, Tonini G, Dammacco F, Zanette G, Meschi F, Porzio O, Bottazzo G, Crino A, Lorini R, Cerutti F, Vanelli M, Barbetti F (2005) KCNJ11 Activating mutations in Italian patients with permanent neonatal diabetes. Human Mutation 25:22–27
Yorifuji T, Nagashima K, Kurokawa K, Kawai M, Oishi M, Akazawa Y, Hosokawa M, Yamada Y, Inagaki N, Nakahata T (2005) The C42R mutation in the Kir6.2 (KCNJ11) gene as a cause of transient neonatal diabetes, childhood diabetes, or later-onset, apparently type 2 diabetes mellitus. J Clin Endocrinol Metab 90:3174–3178
Gloyn AL, Reimann F, Girard C, Edghill EL, Proks P, Pearson ER, Temple IK, Mackay DJG, Shield JPH, Freedenberg D, Noyes K, Ellard S, Ashcroft FM, Gribble FM, Hattersley AT (2005) Moderately Activating Mutations in KCNJ11 may result in relapsing diabetes. Hum Mol Genet 14:925–934
Temple IK, Gardner RJ, Mackay DJ, Barber JC, Robinson DO, Shield JP (2000) Transient neonatal diabetes: widening the understanding of the etiopathogenesis of diabetes. Diabetes 49:1359–1366
Sakura H, Ammala C, Smith PA, Gribble FM, Ashcroft FM (1995) Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic β-cells, brain, heart and skeletal muscle. FEBS Lett 77:338–344
Inagaki N, Gonoi T, Clement IV JP, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J (2005) Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270:1166–1169
Proks P, Arnold A, Bruining J, Girard C, Flanagan SE, Larkin B, Colclough K, Hattersley AT, Ashcroft FM, Ellard SE (2006) A heterozygous activating mutation in the sulphonylurea receptor SUR1 (ABCC8) causes neonatal diabetes. Hum Mol Genet 15:1793–1800
Shyng S, Nichols CG (1997) Octameric stoichiometry of the KATP channel complex. J Gen Physiol 110:655–664
Mikhailov MV, Campbell JD, de Wet H, Shimomura K, Zadek B, Collins RF, Sansom MSP, Ford RC, Ashcroft FM (2005) 3-D structural and functional characterization of the purified KATP channel complex Kir6.2-SUR1. EMBO J 24:4166–4175
Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM (1997) Truncation of Kir6.2 produces ATP-sensitive K-channels in the absence of the sulphonylurea receptor. Nature 387:179–183
Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP 4th, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J (1996) Adenosine diphosphate as an intracellular regulator of insulin secretion. Science 272:1785–1787
Gribble FM, Tucker SJ, Haug T, Ashcroft FM (1998) MgATP activates the β-cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci U S A 95:7185–7190
Ashcroft FM, Rorsman P (1989) Electrophysiology of the pancreatic β-cell. Prog Biophys Mol Biol 54:87–143
Ashcroft FM, Rorsman P (2004) Type-2 diabetes mellitus: not quite exciting enough? Hum Mol Genet 13:R21–R31
Proks P, Antcliff JF, Lippiat J, Gloyn A, Hattersley AT, Ashcroft FM (2004) Molecular basis of Kir6.2 mutations associated with neonatal diabetes or neonatal diabetes plus neurological features. Proc Natl Acad Sci USA 101:17539–17544
Proks P, Girard C, Haider S, Gloyn AL, Hattersley AT, Sansom MSP, Ashcroft FM (2005) A novel gating mutation at the internal mouth of the Kir6.2 pore is associated with DEND syndrome. EMBO Rep 6:470–475
Proks P, Girard C, Ashcroft FM (2005) Enhanced activation by MgATP contributes significantly to the functional effects of KCNJ11 mutations causing neonatal diabetes. Hum Mol Genet 14:2717–26
Tammaro P, Girard C, Molnes J, Njølstad PR, Ashcroft FM (2005) Kir6.2 mutations causing neonatal diabetes provide new insights into Kir6.2-SUR1 interactions. EMBO J 24:2318–2330
Shimomura K, Girard CAJ, Proks P, Nazim J, Lippiat JD, Cerutti F, Lorini R, Ellard S, Hattersely A, Barbetti F, Ashcroft FM (2006) Mutations at the same residue (R50) of Kir6.2 (KCNJ11) that cause neonatal diabetes produce different functional effects. Diabetes 55:1705–1712
Proks P, Girard G, Bævre H, Njølstad P, Ashcroft FM (2006) Clinical and functional effects of a mutation, F35V, in the N-terminus of Kir6.2 (KCNJ11) causing neonatal diabetes. Diabetes 55:1731–1737
Gribble FM, Reimann F (2003) Sulphonylurea action revisited: the post-cloning era. Diabetologia 46:875–891
Zung A, Glaser B, Nimri R, Zadik Z (2004) Glibenclamide treatment in permanent neonatal diabetes mellitus due to an activating mutation in Kir6.2. J Clin Endocrinol Metab 89:5504–5507
Gribble FM, Ashfield R, Ämmälä C, Ashcroft FM (1997) Properties of cloned ATP-sensitive K-currents expressed in Xenopus oocytes. J Physiol 498:87–98
Antcliff JF, Haider S, Proks P, Sansom MSP, Ashcroft FM (2005) Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J 24:229–239
Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA (2003) Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300:1922–1926
John SA, Weiss JN, Xie LH, Ribalet B (2003) Molecular mechanism for ATP-dependent closure of the K+ channel Kir6.2. J Physiol 552:23–34
John SA, Weiss JN, Ribalet B (2005) ATP Sensitivity of ATP-sensitive K channels. Role of the ATP’s γ Phosphate Group and the R50 residue of Kir6.2. J Physiol 568(Pt 3):931–940
Proks P, Gribble FM, Adhikari R, Tucker SJ, Ashcroft FM (1998) Involvement of the N-terminus of Kir6.2 in the inhibition of the KATP channel by ATP. J Physiol 514.1:19–25
Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Slesinger PA, Choe S (2005) Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci 8:279–287
Lin YW, Jia T, Weinsoft AM, Shyng SL (2003) Stabilization of the activity of ATP-sensitive potassium channels by ion pairs formed between adjacent Kir6.2 subunits. J Gen Physiol 122:225–237
Trapp S, Proks P, Tucker SJ, Ashcroft FM (1998) Molecular analysis of KATP channel gating and implications for channel inhibition by ATP. J Gen Physiol 112:333–349
Enkvetchakul D, Loussouarn G, Makhina E, Nichols C (2001) ATP interaction with the open state of the KATP channel. Biophys J 80:719–728
Koster JC, Marshall BA, Ensor N, Corbett JA, Nichols CG (2000) Targeted overactivity of beta cell KATP channels induces profound neonatal diabetes. Cell 100:645–654
Pearson ER, Flechtner I, Njølståd PR, Neonatal Diabetes International Collaborative Group, Malecki MT, Flanagan S, Larkin B, Ashcroft FM, Klimes I, Codner E, Iotova V, Slingerland AS, Shield J, Robert JJ, Holst JJ, Clark PM, Ellard S, Sovik O, Polak M, Hattersley AT (2006) The successful transfer of insulin treated patients with diabetes due to Kir6.2 (KCNJ11) mutations to sulphonylurea tablets. New Eng J Med 355:467–477
Acknowledgements
We thank C. Luzuriaga, J.M. Gomez Vida, A. Aragones, C. Fernandez, R. Barrio, J.P. Lopez-Siguero, and J. Prieto for informing us of patients with neonatal diabetes, and Vicky Ball and Itziar Estalella for technical assistance. FMA thanks the Wellcome Trust, the Royal Society and the EU (EuroDia) for support. The Spanish group was partially supported by grants RGDM (G03/212) and RCMN (C03/08) from the Instituto de Salud Carlos III, Madrid, Spain. GPN is a FIS Research Scientist supported by the Spanish Ministry of Health (Fellowship no. 03/0064). FMA is a Royal Society Research Professor.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Girard, C.A.J., Shimomura, K., Proks, P. et al. Functional analysis of six Kir6.2 (KCNJ11) mutations causing neonatal diabetes. Pflugers Arch - Eur J Physiol 453, 323–332 (2006). https://doi.org/10.1007/s00424-006-0112-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-006-0112-3