
Abstract
Purpose
PGG–PTX is a water-soluble formulation of paclitaxel (PTX), made by conjugating PTX to poly(l-γ-glutamylglutamine) acid (PGG) via ester bonds, that spontaneously forms a nanoparticle in aqueous environments. The purpose of this study was to compare the pharmacokinetics and tissue distribution of PTX following injection of either free PTX or PGG–PTX in mice.
Experimental design
Both [3H]PTX and PGG–[3H]PTX were administered as an IV bolus injection to mice bearing SC NCI-H460 lung cancer xenografts at a dose of 40-mg PTX equivalents/kg. Plasma, tumor, major organs, urine, and feces were collected at intervals out to 340 h. Total taxanes, taxane extractable into ethyl acetate, and native PTX were quantified by liquid scintillation counting and HPLC.
Results
Conjugation of PTX to the PGG polymer increased plasma and tumor C max, prolonged plasma half-life and the period of accumulation in tumor, and reduced washout from tumor. In plasma injection of PGG–PTX increased total taxane AUC0–340 by 23-fold above that attained with PTX. In tumors, it increased the total taxane by a factor of 7.7, extractable taxane by 5.7, and native PTX by a factor of 3.5-fold. Conjugation delayed and reduced total urinary and fecal excretion of total taxanes.
Conclusions
Incorporation of PTX into the PGG–PTX polymer significantly prolonged the half-life of total taxanes, extractable taxane, and native PTX in both the plasma and tumor compartments. This resulted in a large increase in the amount of active PTX delivered to the tumor. PGG–PTX is an attractive candidate for further development.
Similar content being viewed by others

Introduction
Paclitaxel (PTX) has significant anti-tumor activity in patients with ovarian, breast, head, and neck cancer, and non-small-cell lung carcinomas as well as sarcomas [10, 22]. Since PTX has limited solubility in water, it is currently formulated in a solution containing 6-mg PTX/ml in Cremophor EL and ethanol (50% v/v) that must be further diluted before administration. Cremaphor EL is a pharmacologically active compound and its use is associated with acute hypersensitivity reactions [5, 14]. Numerous attempts have been made to develop PTX formulations with reduced systemic toxicity and an enhanced therapeutic index using other vehicles. These attempts have included the use of liposomes, microspheres, micelles, nanoparticles, prodrugs, and polymer–drug conjugates [5, 9, 11, 24, 27]. The greatest success achieved to date has been with paclitaxel protein-bound particles for injection (Abraxane) which is now marketed for the treatment of breast cancer.
In a continuing attempt to further improve the tumor targeting and efficacy of PTX, PTX was covalently conjugated to poly-(L-glutamic acid) (PG–PTX) to produce paclitaxel poliglumex which has been in clinical development for some time. This conjugate is less toxic than PTX and has been reported to have significant antitumor activity in a variety of preclinical models [3, 6, 10, 17, 26, 28]. Pharmacokinetic studies in mice and patients indicate that PG–PTX has a much longer plasma residence time than PTX, and only a small amount of free PTX is present in the plasma after IV injection of PG–PTX (reviewed in [27]). However, despite favorable phase II clinical trial results [12, 21, 23], three randomized phase III trials in patients with non-small cell lung cancer failed to demonstrate an improvement in either progression-free or overall survival [8, 16, 18].
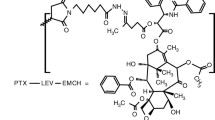
Poly(l-γ-glutamylglutamine) (PGG) is a novel type of polymer consisting of a poly-glutamate in which an additional glutamine side chain has been added to each glutamyl monomer in the backbone as shown in Fig. 1. When the additional glutamines are added to 70 kDa PGA, and PTX is covalently conjugated via an ester linkage to 35% (w/w), the PGG–PTX spontaneously forms a micellar nanoparticle in aqueous solutions with a median diameter of 20 nm as determined by dynamic light scattering. This novel formulation has activity superior to that of Abraxane in the B16 murine melanoma, NCI-H460 non-small cell lung cancer, and 2008 ovarian cancer models. The goal of the present study is to determine how conjugation of PTX to the PGG polymer backbone alters the pharmacokinetics, tissue distribution, and excretion of PTX in nu/nu mice bearing NCI-H460 human lung cancer xenografts.

Structure of PGG–PTX, a random ester conjugate of poly(l-γ-glutamylglutamine) and paclitaxel. The structure shown is illustrative of a fragment of the molecule, but specific conjugation sites are not implied. There are approximately five non-conjugated monomer glutamylglutamine units per paclitaxel-conjugated monomer glutamylglutamine unit
Materials and methods
Drugs
Paclitaxel (PTX) was obtained from Nublock, LLC. Vista, CA. [3H]PTX was purchased from Moravek Biochemicals, Inc. (Brea, CA) and had a specific activity of 1 mCi/177 μg PTX (or 56.50 mCi/mg PTX). Unlabeled PTX was dissolved in ethanol and mixed with a stock solution of [3H]PTX to yield [3H]PTX with a specific activity of 9.98 μCi/mg. After evaporation of the ethanol, [3H]PTX was redissolved in Cremophor/alcohol (1:1 v/v) at a concentration of 30 mg/ml. This solution was further diluted 1:5 in 0.9% NaCl in water to produce a final concentration of 6.0 mg/ml prior to IV injection. PGG–[3H]PTX was synthesized by conjugating [3H]PTX to 70 kDa poly(l-glutamic acid) in which each glutamyl monomer was substituted with an additional glutamine and then loaded to 35% w/w with PTX and [3H]PTX to produce a final specific activity of 9.98 μCi/mg equivalent PTX. Before injection, the conjugate was dissolved in saline to an equivalent PTX concentration of 6 mg/ml and filtered through a 0.22-μm sterile filter.
Animals
Female nu/nu mice (22–30 g) were obtained from Charles River Laboratories and maintained in a pathogen-free vivarium. All experiments involving animals were performed in accordance with the guidelines of the institution’s Animal Care and Use Committee. Mice were inoculated SC with 4 × 106 NCI-H460 human lung cancer cells that had been grown in tissue culture on each shoulder and hip. At the point when the mean tumor volume for the entire population had reached 400–500 mm3, each mouse received a single IV bolus injection of [3H]PTX or PGG–[3H]PTX at a dose of 40 mg PTX equivalents/kg.
Sample collection and counting of total drug radioactivity
Six mice were anesthetized at each of 0, 0.166, 0.5, 2, 4, 24, 48, 72, 144, 240, and 340 h after injection of either [3H]PTX or PGG–[3H]PTX and at least 0.5 ml of whole blood was drawn from the heart immediately prior to killing and after induction of anesthesia with ketamine 100 mg/kg and xylacine 10 mg/kg. The blood was placed in an Eppendorf tube containing 10 μl heparin 1,000 U/ml and plasma was separated from formed elements by centrifugation and frozen for later processing. Each of the four tumors per mouse and a ~100 mg sample from the right lower lobe of the lung, right lobe of the liver, right kidney, and skeletal muscle, and the entire spleen was dissected free of surrounding tissue, blotted free of fluid, and weighed. The tissue samples were then homogenized in 1.0 ml of phosphate buffered saline, pH 7.4, using a Polytron PT1035 (Kinematica, Lucerne, Switzerland). One hundred microliters of each sample was then added to the scintillation vials with 4.9-ml scintillation solution and stored for 2 days in the dark to allow any chemiluminescence to subside. The radioactivity was then quantified on a scintillation counter (Beckman LS 6000 LL, Beckman Instruments, Fullerton, CA), and content determined from control curves in which drug was added directly to the homogenates made from mice not injected with any drug.
Extraction of drug from plasma and tumor tissue
Non-polymer bound PTX and hydrophobic PTX metabolites were extracted from 100-μl aliquots of plasma or tissue homogenate by mixing 1.0-ml ethyl acetate for 1 h at room temperature. The sample was then centrifuged at 2,500×g for 10 min, the organic phase was removed and the aqueous phase re-extracted with two additional 1.0-ml aliquots of ethyl acetate. The three organic extracts were then combined, dried, and then resuspended in 100 μl of 50% acetonitrile. Total extractable drug was quantified by scintillation counting as described above.
HPLC assay of extractable native PTX
The fraction of the total extractable [3H] that was still in the form of native PTX following injection of either [3H]PTX or PGG–[3H]PTX was determined by separation of native drug from metabolites by HPLC. A 100-μl aliquot of either plasma or tissue homogenate was extracted with 5 vol of ethyl acetate and the organic phase was then recovered, dried, and the residue redissolved in 95-μl of 50% acetonitrile and mixed with 5-μl non-radiolabeled PTX at a concentration of 0.1 mg/ml. A total of 100 μl of the reconstituted solution was injected onto a Beckman HPLC equipped with an online scintillation detector. The HPLC system consisted of a 150 × 4 mm Phenomenex column, a UV/visible light detector set at 228 nm (System Gold 168 Detector), and a flow scintillation analyzer (PerkinElmer Radiomatic 610 TR). The column was eluted with a linear 20–95% acetonitrile gradient at a flow rate of 0.3 ml/min for 30 min, a 30 min washout was allowed for the system to return to initial conditions. The retention time of native paclitaxel was 20 min, the offset time between the UV and radioactive detectors was ~0.2 min, the efficiency of [3H] counting was 33%. The radioactivity migrating with the peak of native PTX was quantified. A standard curve was established for plasma and each type of tissue homogenate separately by adding known amounts of [3H]PTX to 1 ml of plasma or tissue homogenates from mice not injected with any radioactivity and the drug content was expressed as counts per minute per gram of tissue or milliliter plasma. The standard curves were linear from 45 to 3,000 ng/ml and were run with each set of tissue extracts. The lower limit of quantitation was 45 ng/ml.
Determination of drug excretion and elimination
To determine the routes of elimination of [3H]PTX following injection of either [3H]PTX or PGG–[3H]PTX, normal female nu/nu mice were injected with [3H]-PTX (6 mice) or PGG–[3H]PTX (6 mice) at a dose of 40 mg PTX equivalents/kg body weight. The mice were placed in metabolic cages, and urine and feces were collected during the following intervals: 0–4, 4–8, 8–24, 24–48, 48–72, 72–96, 96–120, 120–144, 144–168, 168–196, 196–210 and 210–240 h. The collected samples were analyzed for total radioactivity.
Estimation of pharmacokinetic parameters
Pharmacokinetic analysis was based on non-compartmental methods using WinNolin version 5.2 (Pharsight Corporation, San Francisco, CA, USA). The area under the drug concentration–time curves was calculated from mean tissue content values observed from the time of drug injection to 340 h after administration using the linear/log trapezoidal rule. The curves shown in the figures represent mean values as a function of time and are not fitted to the data.
Statistics
All two-way comparisons were made with Student’s t test assuming unequal variance of the samples. A P value of <0.05 was considered significant.
Results
PGG–PTX stability
The rate of release of PTX from the PGG polymer backbone was examined by incubating PGG–PTX at a concentration of 6 mg/ml in vitro in fresh human plasma at 37°C. The free native PTX was then quantified by HPLC analysis following extraction into ethyl acetate. Figure 2 shows that there was no detectable immediate release of PTX. The rate of release was more rapid over the first 6 h and then slowed after the first 10 h and was subsequently quite constant till 144 h. A total of 6.1 ± 1.0 (SEM) % of the PTX was released in 24 h.

Release of PTX from PGG–PTX as a function of time during incubation in fresh human plasma at 37°C. Each point represents the mean of three samples; vertical bars, SEM
Pharmacokinetics of total taxanes, extractable taxanes, and native PTX in plasma
To determine the pharmacokinetic profile of PGG–PTX relative to that of PTX in plasma and tissues, female nu/nu mice bearing subcutaneous NCI-H460 human lung cancer xenografts were given IV bolus doses of either [3H]PTX or PGG–[3H]PTX at a dose of 40-mg PTX equivalents/kg. Samples of plasma, tumor, and the major organs were collected at various time points till 340 h after the injection and the levels of three different forms of the drugs were determined by scintillation counting. The level of total taxane was determined by measuring the radioactivity in the entire sample which included PTX bound to plasma proteins or the PGG polymer backbone, as well as native drug and metabolites. When the plasma or tissue homogenates were extracted with ethyl acetate, the water-soluble PGG–[3H]PTX remained in the aqueous phase while native PTX with any metabolites, referred to here as ‘extractable taxanes’, were recovered in the organic phase. Since the relative activities of the components in the ‘extractable taxanes’ are unknown, native PTX was separated from its metabolites in the ethyl acetate extract on an HPLC column which permitted quantification of the native form of PTX present at each time point following injection of either [3H]PTX or PGG–[3H]PTX [2, 10, 26, 27].
As shown in Fig. 3, and summarized in Table 1, there were substantial differences in the plasma pharmacokinetics of PTX and PGG–PTX. The total taxane C max was 8.5-times higher in animals given PGG–PTX than in those given PTX. The estimated terminal half-life values for total taxanes were 293.6 h for PGG–PTX and 59.9 h for PTX. As shown graphically in Fig. 3c, the AUC0–340 of total taxanes in mice injected with PGG–PTX was 23.6-fold greater than that for mice injected with PTX. Consistent with the polymeric structure of PGG–PTX, its volume of distribution was only 21%, and its plasma clearance was only 4%, of that of PTX.

Concentration of total taxane, extractable taxane and native PTX in plasma as a function of time following injection of PTX or PGG–PTX. a Following injection of PTX plotted over 0–340 h, b following injection of PTX plotted over just the first 24 h, c following injection of PGG–PTX plotted over 0–340 h, d following injection of PGG–PTX plotted over just the first 24 h, e the AUC0–340 values following injection of PGG–PTX relative to the following injection of PTX. Filled square total taxane, filled diamond extractable taxane, filled triangle native PTX. Each data point is the mean of six samples obtained from six mice at each time point; vertical bars, SEM
There were more modest differences in extractable taxane and native PTX levels following injection of PTX or PGG–PTX. The C max and AUC0–340 of extractable taxane produced by injection of PGG–PTX were only 1.2 and 4.9-fold, respectively, higher than that produced by injection of PTX. Likewise, the C max and AUC of native PTX produced by injection of PGG–PTX were only 1.5- and 4.0-fold, respectively, higher than that produced by injection of PTX. These results are consistent with the concept that PTX is released from PGG–PTX quite slowly in plasma, and that the larger AUC for extractable taxane and native PTX following injection of PGG–PTX is largely due to its more prolonged half-life.
Pharmacokinetics of total taxanes, extractable taxanes, and native PTX in tumor
Figure 4 presents the curves of tumor content of each of the three forms of the drugs as a function of time following injection of either PGG–PTX or PTX, and the estimated pharmacokinetic parameters are presented in Table 2.

Content of total taxane, extractable taxane, and native PTX in tumor as a function of time following injection of PTX or PGG–PTX. a Following injection of PTX plotted over 0–340 h, b following injection of PTX plotted over just the first 24 h, c following injection of PGG–PTX plotted over 0–340 h, d following injection of PGG–PTX plotted over just the first 24 h, e the AUC values following injection of PGG–PTX relative to the following injection of PTX. Filled square total taxane, filled diamond extractable taxane, filled triangle native PTX. Each data point is the mean of samples obtained from 24 independent tumors; vertical bars, SEM
Comparison of tumor to plasma for PGG–PTX
In contrast to the situation in plasma, following injection of PGG–PTX, the peak tumor concentration of total taxane was only 2.1-fold higher than that of extractable taxane and 4.3-fold higher than that of native PTX indicating that a larger fraction of the total drug present at the time of the peak was in the form of native drug in the tumor tissue than in plasma. The content of the extractable taxane and native PTX initially declined more rapidly than the total taxane level. As shown in Fig. 4c, the AUC0–340 for native PTX over the time period measured was fully 20% of that of the total taxane (compared to only 3.6% for plasma) and that for extractable taxane was 32% of the total (compared to 8.1% for plasma). This result indicates that relatively more of the PTX came off from the polymer backbone and/or was metabolized to ethyl acetate-extractable forms during the first 340 h after PGG–PTX injection in tumor than in plasma. Overall, the total exposure of the tumor to total taxane was only 72% for plasma; however, exposure of the tumor to extractable taxane was 2.9-fold higher than in plasma and exposure to native PTX was 4.0-fold higher. This result indicates that PGG–PTX delivered more exposure to native PTX to the tumor than to the plasma, indicative of its ability to concentrate in the tumor.
Comparison of tumor to plasma for PTX
In mice injected with PTX, the peak tumor concentration of total taxane in the tumor was only 1.4-fold higher than that of extractable taxane (compared to 2.3-fold higher for plasma) and only 1.5-fold higher than that of native PTX (compared to 5.2-fold higher in plasma). At the peak time, 66% fully of the total level was made up of native drug. This suggests that, relative to native drug in plasma, native PTX was concentrated into the tumor. The AUC0–340 for native PTX over the time period measured was 45% of that of the total taxane and that for extractable taxane was 43% of the total, both values were higher than that observed in plasma (21 and 39%, respectively). To summarize, the exposure to native drug for the tumor was 14% greater than that for the plasma (Figs. 3c, 4c). In contrast to the situation following injection of PGG–PTX, the decline in the levels of extractable taxane and native PTX in the tumor was only marginally more rapid than that of the total taxane.
Drug levels produced by PTX versus PGG–PTX in tumor
As for the plasma pharmacokinetics, the behavior of the two drugs was quite different with respect to accumulation and washout from the tumor tissue. The key differences were that, while the peak concentration of total taxane attained in the tumor (C max) after injection of PGG–PTX was 2.1-fold higher than after injection of PTX, the total taxane level in the tumor following injection of PGG–PTX continued to increase for a 1.9-fold longer period of time following injection of PTX. The time of maximal accumulation for PGG–PTX was 4 h whereas it was just 2 h for PTX. In addition, the washout of PGG–PTX was slower as reflected by the 1.9-fold longer estimated terminal half-life. The combination of a higher C max, a more prolonged tumor accumulation phase, and slower washout resulted in an AUC0–340 for total taxane in the tumor being 7.7-fold greater for PGG–PTX than for PTX (Figs. 3c, 4c).
The C max of extractable taxanes in the tumor was 1.4-fold higher following injection of PGG–PTX than PTX but the main difference was that the extractable taxane generated by PGG–PTX continued to accumulate in the tumor for a much longer period of time (12-fold) such that the time of maximal accumulation was 24 h versus just 2 h following injection of PTX. The combination of a higher C max, more prolonged tumor accumulation phase, and slower washout resulted in a total exposure for the tumor being 5.7-fold greater for extractable taxanes following injection of PGG–PTX than of PTX.
Assuming that the native drug is the most important with respect to tumor cell kill, a comparison of the AUC0–340 for native drug in mice injected with PGG–PTX to that in mice injected with PTX is of particular interest. The C max produced by the former was 1.4-fold higher than that produced by the latter, but there was a large difference in the period of accumulation (12-fold) and washout (3.4-fold) in favor of PGG–PTX. This resulted in an AUC0–340 for native drug that was 3.5-fold higher following injection of PGG–PTX. Thus, PGG–PTX was substantially more effective at delivering the most important form of the drug to the tumor than PTX.
Pharmacokinetics of total taxanes in major organs
Whereas, all three forms of the drugs were measured in the plasma and tumor, only total taxanes were measured in the major organs. The drug content per gram wet weight is shown for liver, lung, kidney, spleen, and skeletal muscle as a function of time in Fig. 5. The estimated pharmacokinetic parameters are presented in Table 3. The data for plasma and tumor are included in Table 3 for ease of comparison, and Fig. 6 presents a summary of the C max and AUC data in graphic form.

Content of total taxane in liver, lungs, kidneys, spleen, and skeletal muscle as a function of time following injection of PTX (filled triangle) or PGG–PTX (filled circle). Each data point is the mean of six independent samples; vertical bars, SEM

Comparison of the C max and AUC of total taxane produced in major organs by injection of either PGG–PTX or PTX. C max and AUC are the ratio of levels produced by PGG–PTX thanthose produced by PTX. Vertical bars, SEM
In all the organs tested, decay in the content of total taxane following injection of PGG–PTX was much slower than PTX. PGG–PTX produced C max values of total taxane that were quite similar in the liver, lungs, and spleen whereas they were more variable following injection of PTX with a particularly high level in the lung. Given the particulate nature of the PGG–PTX, one might have expected a higher C max following injection of PGG–PTX in both the liver and spleen; however, while they were 2.7-fold higher in the spleen, the liver levels were similar. The levels in the liver were also of interest because, the total taxane content dropped rapidly between 0.166 and 48 h after injection of PTX; following injection of PGG–PTX, the level continued to increase over this interval eventually reaching a peak concentration equal to 90% of that attained by PTX. Once having reached its peak level, the washout of total taxane generated by PGG–PTX was very slow; the clearance from the liver after injection of PGG–PTX was only 1.5% of that after administration of PTX. The estimated terminal half-life of total taxane washout from the liver was 9.5-fold slower for PGG–PTX than for PTX resulting in 33.4-fold AUC0–340 for PGG–PTX as shown in Fig. 6. The drugs also exhibited quite different behavior in the lungs. The C max produced by PGG–PTX was only 60% of that produced by PTX; however, the AUC produced by PGG–PTX was 34.5-fold higher than that produced by injection of PTX and the estimated terminal half-life of total taxane washout from the lung was 3.4-fold slower for PGG–PTX than PTX. The clearance of total taxane from the lungs following injection of PGG–PTX was only 2.5% of that after injection of PTX.
The pattern of accumulation and washout of total taxane in the kidneys resembled that of the liver. Whereas, the level of total taxane following injection of PTX started falling right away, after administration of PGG–PTX, it continued to accumulate over the first 1 h and thereafter the level remained quite stable, such that the concentration at the last time point measured was the same as the C max. The peak kidney level associated with PGG–PTX was only 90% of that produced by PTX but the total exposure was 30.9-fold higher. The very slow washout of PGG–PTX from the kidney precluded estimation of the terminal half-life or clearance.
The pattern of accumulation and washout of total taxane produced by PGG–PTX and PTX in the spleen again resembled that in other organs with the exception that the C max after injection of PGG–PTX exceeded that produced with PTX by 2.7-fold. Whereas, the level declined rapidly after injection of PTX, it continued to accumulate over the first 4 h after administration of PGG–PTX and thereafter remained quite stable. The total exposure was 72.8-fold higher for PGG–PTX and PTX. The very slow washout from the spleen after injection of PGG–PTX precluded estimation of the terminal half-life or clearance.
Muscle was the only tissue in which the pattern of accumulation and washout differed significantly from that in liver, lungs, kidneys, and spleen. In muscle, total taxane after injection of PGG–PTX did not show a period of accumulation as was seen in all the other tissues; instead, levels followed plasma levels, progressively declining after completion of the injection. In addition, the total taxane and AUC ratio of 2.7 was substantially smaller than that observed for the other tissues suggesting that PGG–PTX nanoparticles have a lower affinity for muscle than the other tissues tested.
Excretion of total taxanes derived from PTX and PGG–PTX in the urine and feces
Groups of six mice injected with 40-mg PTX equivalents/kg of either [3H]PTX or PGG–[3H]PTX were maintained in metabolic cages, and urine and feces were quantitatively recovered over a period of 240 h. The total taxane level was measured by scintillation counting with appropriate controls for possible quenching. As shown in Fig. 7, the two drugs showed quite different patterns of urinary excretion. The urinary excretion of total taxanes following injection of PTX was more rapid than that after injection of PGG–PTX over the first 24 h. Following injection of PGG–PTX, 13.3% of the PTX dose administered was excreted in urine within the first 48 h and 16.9% over the entire 240 h. In contrast, following injection of PTX, 23.4% was excreted in the urine within the first 48 h and 24.7% was excreted in 240 h. Thus, essentially all the radioactivity excretable in the urine following the injection of unconjugated PTX appeared in the first 24 h and very little thereafter. In contrast, while most of the radioactivity excretable in the urine following injection of PGG–PTX came out in the first 48 h, an additional 3% appeared in the urine at later time points.

Urinary and fecal excretion of PTX as a function of time after injection of PGG–PTX
A substantial fraction of the administered dose was recovered in feces for both drugs. The fecal excretion of total taxane following injection of PTX was substantially more rapid than that of PGG–PTX over the first 24 h. Following injection of PGG–PTX, 36.5% of the injected dose was excreted in the feces within the first 48 h and 45.3% over the entire 240-h. In contrast, following injection of PTX, 72.3% was excreted in the feces within the first 48 h and 95.2% was excreted over the full 240 h sampling period. Thus, the majority of the radioactivity excreted in the feces following injection of PTX came out in the first 48 h whereas most of the radioactivity excreted in the feces following injection of PGG–PTX came out in the first 48 h, an additional 23% appeared in the feces at later time points.
Discussion
Polymer–drug conjugates have been investigated as carriers for anticancer drugs in an attempt to direct active agents to tumors in vivo and to reduce toxic effects to normal tissues [4, 7, 10, 15]. The increased antitumor efficacy of polymeric drugs has been shown to be largely attributable to the enhanced permeability and retention (EPR) effect of macromolecules [15]. At present, a few polymer–drug conjugates have reached the stage of clinical application. A hydroxypropyl methacrylamide copolymer (HPMA)–doxorubicin conjugate (PK1) was shown to have antitumor activity and decreased toxicity relative to free doxorubicin in patients with refractory tumors in phase I clinical studies [30] but was not developed further. HPMA–cisplatin and HPMA–DACH platinum conjugates have demonstrated substantial activity in preclinical studies [13, 20] and favorable pharmacokinetics in initial phase 1 clinical trials [19]. Paclitaxel poliglumex, a formulation in which paclitaxel is linked to a PGA polymer, has activity in preclinical studies and early clinical trials but has yet to demonstrate a superior therapeutic index in randomized phase III trials.
PGG–PTX differs significantly from paclitaxel poliglumex in two important ways. First, while the molecular weight of the polymer in paclitaxel poliglumex is 35 kDa by viscosity and ~20 kDa by multi-angle light scattering, the addition of a glutamine acid to each glutamyl in the 70-kDa polymer backbone of PGG increases its molecular weight to the range of ~52 kDa by multi-angle light scattering. Second, when PGG is loaded to 35% by weight with PTX, it forms micelles with a median diameter of 20 nm in aqueous environments and thus can be expected to behave more as a nanoparticle than as a flexible polymer as it traverses the circulation and tumor extracellular compartments. PGG–PTX was designed to deliver more PTX to the tumor than to other dose-limiting tissues. In order to achieved this, a formulation was needed that would release little of its PTX cargo in the systemic circulation, but would release essentially all of it once in the tumor. Although the PTX is linked to the PGG backbone via an ester linkage, the rate of release of PTX from the polymer in human plasma was quite slow and only 6.1% was released within 24 h. Although the rate of release of PTX from the polymer could not be measured directly in the tumor, the goal of increasing delivery of PTX to the tumor was clearly achieved as documented by increased total taxane, extractable taxane, and native PTX exposures for the tumor following injection of PGG–PTX compared to administration of an equal amount of unconjugated PTX. The AUC ratios were 7.7 for total taxane, 5.7 for extractable taxane, and 3.5 for native PTX. These increases were the result of both modestly higher C max values but substantially more prolonged periods of drug accumulation in the tumor and slower washout of all three forms of the drug following administration of PGG–PTX compared to PTX.
One of the questions of interest with regard to PGG–PTX is whether the increase in tumor exposure is simply the result of an increase in plasma AUC, or whether the PGG–PTX micelles are truly able to accumulate in the tumor. The observation that the tumor AUC for all three forms of the drug was much higher than the plasma AUC indicates that a large degree of targeting was in fact attained even when taking into consideration that the units for the plasma are μg h/ml whereas those for the tumor are μg h/g. The ratio was 17 for total taxane, 14 for extractable taxane, and 16 for native PTX.
As shown in Fig. 5, PGG–PTX delivered a lot more total taxane to the liver, lungs, kidneys, and spleen than to the tumor. However, it is important to note that the clinical efficacy of PTX is based not on the fact that most drug gets to the tumor than to other tissues, but that more of it gets into the tumor than to those tissues that limit its dose. None of these organs are dose-limiting for PTX. Despite increasing exposure for the liver, lungs, spleen, and kidneys by large amounts, in mice PGG–PTX is substantially less toxic than PTX. In nu/nu mice, the maximum tolerated dose of a single injection of PGG–PTX is ~400 mg/kg whereas the maximum tolerated dose of a single IV injection of PTX is 100 mg/kg. Thus, the expectation is that, when injected at equitoxic doses instead of equimolar doses, the magnitude of the increase in the amount of drug delivered to the tumor will be even greater.
Consistent with studies of paclitaxel poliglumex [25], linkage of PTX to the PGG backbone markedly altered its urinary and fecal excretion. Whereas, excretion of taxanes derived from injection of unconjugated PTX was largely complete in the urine in 24 h, and in the feces by 120 h, and a total of 95.7% of the injected dose had been excreted by both routes in 240 h the excretion of taxanes after the injection of PGG–PTX continued throughout the entire sampling period and only 49.8% of the total dose had been excreted by 240 h. Thus, linkage of PTX to PGG markedly reduced its access to both the urinary and biliary excretion pathways and prolonged its residence time. It is important to note that only total radioactivity was measured in this experiment and it remains unknown what fraction of the radioactivity appearing in either the urine or feces is unchanged PTX or PGG–PTX and how much is metabolites. The data from these experiments are nevertheless consistent with the previous findings that hepatic metabolism and biliary excretion are principal mechanisms of PTX elimination [1, 29]. PGG–PTX appears to have less relative access to hepatic metabolism and therefore has a lower fecal excretion. The data from this experiment are consistent with the concept that PTX is not released rapidly from the polymer backbone following IV injection. If the PTX had been rapidly released, one would have expected to observe less of a difference in the rate of appearance of radioactivity in the urine and feces compared to unconjugated PTX.
In summary, this study showed that PGG–PTX delivered substantially more of all three forms of PTX to the tumor than did an equivalent PTX dose of 40 mg/kg. Furthermore, the native PTX was present in tumor tissues for up to 340 h after injection of PGG–PTX. The enhanced tumor uptake of PGG–PTX and prolonged release of free PTX within the tumor is consistent with the superior antitumor activity of PGG–PTX observed in preclinical studies.
Abbreviations
- PGA:
-
Poly(l-glutamic acid)
- PGG:
-
Poly(l-γ-glutamylglutamine)
- PGG–PTX:
-
70 kDa PGA to which both additional glutamine side chains and PTX have been added
- PTX:
-
Paclitaxel
References
Bardelmeijer HA, Oomen IA, Hillebrand MJ, Beijnen JH, Schellens JH, van Tellingen O (2003) Metabolism of paclitaxel in mice. Anticancer Drugs 14:203
Briasoulis E, Karavasilis V, Tzamakou E, Rammou D, Soulti K, Piperidou C, Pavlidis N (2004) Interaction pharmacokinetics of pegylated liposomal doxorubicin (Caelyx) on coadministration with paclitaxel or docetaxel. Cancer Chemother Pharmacol 53:452
Calvo P, Gouritin B, Chacun H, Desmaele D, D Angelo J, Noel JP, Georgin D, Fattal E, Andreux JP, Couvreur P (2001) Long-circulating PEGylated polycyanoacrylate nanoparticles as new drug carrier for brain delivery. Pharm Res 18:1157
de Jonge ME, van den Bongard HJ, Huitema AD, Mathot RA, Rosing H, Baas P, van Zandwijk N, Beijnen JH, Schellens JH (2004) Bayesian pharmacokinetically guided dosing of paclitaxel in patients with non-small cell lung cancer. Clin Cancer Res 10:2237
Gelderblom H, Verweij J, Nooter K, Sparreboom A (2001) Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer 37:1590
Kim SC, Yu J, Lee JW, Park ES, Chi SC (2005) Sensitive HPLC method for quantitation of paclitaxel (Genexol in biological samples with application to preclinical pharmacokinetics and biodistribution. J Pharm Biomed Anal 39:170
Kopecek J (1991) Targetable polymeric anticancer drugs Temporal control of drug activity. Ann N Y Acad Sci 618:335
Langer CJ, O’Byrne KJ, Socinski MA, Mikhailov SM, Lesniewski-Kmak K, Smakal M, Ciuleanu TE, Orlov SV, Dediu M, Heigener D, Eisenfeld AJ, Sandalic L, Oldham FB, Singer JW, Ross HJ (2008) Phase III trial comparing paclitaxel poliglumex (CT-2103, PPX) in combination with carboplatin versus standard paclitaxel and carboplatin in the treatment of PS 2 patients with chemotherapy-naive advanced non-small cell lung cancer. J Thorac Oncol 3:623
Li C, Ke S, Wu QP, Tansey W, Hunter N, Buchmiller LM, Milas L, Charnsangavej C, Wallace S (2000) Tumor irradiation enhances the tumor-specific distribution of poly(l-glutamic acid)-conjugated paclitaxel and its antitumor efficacy. Clin Cancer Res 6:2829
Li C, Newman RA, Wu QP, Ke S, Chen W, Hutto T, Kan Z, Brannan MD, Charnsangavej C, Wallace S (2000) Biodistribution of paclitaxel and poly(l-glutamic acid)-paclitaxel conjugate in mice with ovarian OCa-1 tumor. Cancer Chemother Pharmacol 46:416
Li C, Yu DF, Newman RA, Cabral F, Stephens LC, Hunter N, Milas L, Wallace S (1998) Complete regression of well-established tumors using a novel water-soluble poly(l-glutamic acid)-paclitaxel conjugate. Cancer Res 58:2404
Lin NU, Parker LM, Come SE, Burstein HJ, Haldoupis M, Ryabin N, Gelman R, Winer EP, Shulman LN (2007) Phase II study of CT-2103 as first- or second-line chemotherapy in patients with metastatic breast cancer: unexpected incidence of hypersensitivity reactions. Invest New Drugs 25:369
Lin X, Zhang Q, Rice JR, Stewart DR, Nowotnik DP, Howell SB (2004) Improved targeting of platinum chemotherapeutics. The antitumour activity of the HPMA copolymer platinum agent AP5280 in murine tumour models. Eur J Cancer 40:291
Longnecker SM, Donehower RC, Cates AE, Chen TL, Brundrett RB, Grochow LB, Ettinger DS, Colvin M (1987) High-performance liquid chromatographic assay for taxol in human plasma and urine and pharmacokinetics in a phase I trial. Cancer Treat Rep 71:53
Maeda H, Seymour LW, Miyamoto Y (1992) Conjugates of anticancer agents and polymers: advantages of macromolecular therapeutics in vivo. Bioconjug Chem 3:351
O’Brien ME, Socinski MA, Popovich AY, Bondarenko IN, Tomova A, Bilynsky BT, Hotko YS, Ganul VL, Kostinsky IY, Eisenfeld AJ, Sandalic L, Oldham FB, Bandstra B, Sandler AB, Singer JW (2008) Randomized phase III trial comparing single-agent paclitaxel Poliglumex (CT-2103, PPX) with single-agent gemcitabine or vinorelbine for the treatment of PS 2 patients with chemotherapy-naive advanced non-small cell lung cancer. J Thorac Oncol 3:728
Omelyanenko V, Gentry C, Kopeckova P, Kopecek J (1998) HPMA copolymer-anticancer drug-OV-TL16 antibody conjugates. II. Processing in epithelial ovarian carcinoma cells in vitro. Int J Cancer 75:600
Paz-Ares L, Ross H, O’Brien M, Riviere A, Gatzemeier U, Von Pawel J, Kaukel E, Freitag L, Digel W, Bischoff H, Garcia-Campelo R, Iannotti N, Reiterer P, Bover I, Prendiville J, Eisenfeld AJ, Oldham FB, Bandstra B, Singer JW, Bonomi P (2008) Phase III trial comparing paclitaxel poliglumex vs docetaxel in the second-line treatment of non-small-cell lung cancer. Br J Cancer 98:1608
Rademaker-Lakhai JM, Terret C, Howell SB, Baud CM, De Boer RF, Pluim D, Beijnen JH, Schellens JH, Droz JP (2004) A Phase I and pharmacological study of the platinum polymer AP5280 given as an intravenous infusion once every 3 weeks in patients with solid tumors. Clin Cancer Res 10:3386
Rice JR, Gerberich JL, Nowotnik DP, Howell SB (2006) Preclinical efficacy and pharmacokinetics of AP5346, a novel diaminocyclohexane-platinum tumor-targeting drug delivery system. Clin Cancer Res 12:2248
Richards DA, Richards P, Bodkin D, Neubauer MA, Oldham F (2005) Efficacy and safety of paclitaxel poliglumex as first-line chemotherapy in patients at high risk with advanced-stage non-small-cell lung cancer: results of a phase II study. Clin Lung Cancer 7:215
Rowinsky EK, Donehower RC (1995) Paclitaxel (taxol). N Engl J Med 332:1004
Sabbatini P, Aghajanian C, Dizon D, Anderson S, Dupont J, Brown JV, Peters WA, Jacobs A, Mehdi A, Rivkin S, Eisenfeld AJ, Spriggs D (2004) Phase II study of CT-2103 in patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. J Clin Oncol 22:4523
Sharma A, Mayhew E, Straubinger RM (1993) Antitumor effect of taxol-containing liposomes in a taxol-resistant murine tumor model. Cancer Res 53:5877
Singer JW (2005) Paclitaxel poliglumex (XYOTAX, CT-2103): a macromolecular taxane. J Control Release 109:120
Singer JW, Baker B, De Vries P, Kumar A, Shaffer S, Vawter E, Bolton M, Garzone P (2003) Poly-(l)-glutamic acid-paclitaxel (CT-2103) [XYOTAX], a biodegradable polymeric drug conjugate: characterization, preclinical pharmacology, and preliminary clinical data. Adv Exp Med Biol 519:81
Singer JW, Shaffer S, Baker B, Bernareggi A, Stromatt S, Nienstedt D, Besman M (2005) Paclitaxel poliglumex (XYOTAX; CT-2103): an intracellularly targeted taxane. Anticancer Drugs 16:243
Sparreboom A, Scripture CD, Trieu V, Williams PJ, De T, Yang A, Beals B, Figg WD, Hawkins M, Desai N (2005) Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clin Cancer Res 11:4136
Sparreboom A, van Tellingen O, Nooijen WJ, Beijnen JH (1996) Tissue distribution, metabolism and excretion of paclitaxel in mice. Anticancer Drugs 7:78
Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R, Thomson AH, Murray LS, Hilditch TE, Murray T, Burtles S, Fraier D, Frigerio E, Cassidy J (1999) Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents–drug–polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clin Cancer Res 5:83
Acknowledgments
Financial support for this work was provided by UC Discovery grant bio06-10568 and the Nitto Denko Technical Corporation. This work was supported by a public–private grant program operated by the University of California in which the University and the Nitto Denko Technical Corp each provide half of the funding. Drs. Xinghe Wang, Gang Zhao, Sang Van, Nan Jiang and Lei Yu are employees of Nitto Denko. Drs. David Vera and Stephen B. Howell are employees of the University of California, San Diego who receive research support under this grant. Drs. Vera and Howell have also served as consultants to Nitto Denko in the past.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wang, X., Zhao, G., Van, S. et al. Pharmacokinetics and tissue distribution of PGG–paclitaxel, a novel macromolecular formulation of paclitaxel, in nu/nu mice bearing NCI-460 lung cancer xenografts. Cancer Chemother Pharmacol 65, 515–526 (2010). https://doi.org/10.1007/s00280-009-1058-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-009-1058-x