
Abstract
In this work, we present an atomic population study within the Fractional Occupation Hirshfeld-I (FOHI) scheme applied to several pristine, boron-, nitrogen- and B, N-doped non-periodic zigzag graphene nanoribbons (always using a double carbon replacement). To accomplish this task, we have considered the singlet–triplet energy gap as a criterion to check the most reliable electron density. B2PLYPD double-hybrid functional provides the most accurate relative energies in comparison with the other methods, but similar atomic populations are obtained in most cases. Moreover, in spite of the observed different behavior concerning the population of B, N dopants and their corresponding p- and n-type doping effects, the FOHI atomic populations are in excellent agreement with the widely accepted electronegative scale. Nevertheless, we propose to employ a more appropriate electron partitioning strategy taking into account the contribution of \(\pi\)-symmetric orbitals. It provides the expected population results according to doping guidelines. In any case, both kinds of populations describe in a similar way the mesomeric effects and the edge variations after replacing two carbons by either two boron or nitrogen atoms. On the contrary, the populations point out a different behavior when the systems are doped with one boron and one nitrogen simultaneously.





Similar content being viewed by others

Notes
The energies of the considered structures are shown in Table S1 in the electronic supporting information (ESI).
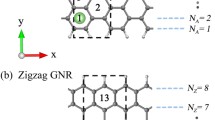
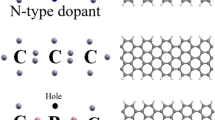
See figures below to understand the numbering.
Unfortunately, the calculation of analytic gradients is not available in Gaussian 09 for ROMP2 and ROB2PLYPD approaches.
References
Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA (2004) Electric field effect in atomically thin carbon films. Science 306(5696):666–669
Terrones M, Botello-Méndez AR, Campos-Delgado J, López-Urías F, Vega-Cantú YI, Rodríguez-Macías FJ, Elías AL, Muñoz Sandoval E, Cano-Márquez AG, Charlier JC, Terrones H (2010) Graphene and graphite nanoribbons: morphology, properties, synthesis, defects and applications. Nano Today 5(4):351–372
Liu H, Liu Y, Zhu D (2011) Chemical doping of graphene. J Mater Chem 21(10):3335–3345
Guo S, Dong S (2011) Graphene nanosheet: synthesis, molecular engineering, thin film, hybrids, and energy and analytical applications. Chem Soc Rev 40(5):2644–2672
Cai J, Ruffieux P, Jaafar R, Bieri M, Braun T, Blankenburg S, Muoth M, Seitsonen AP, Saleh M, Feng X, Mllen K, Fasel R (2010) Atomically precise bottom-up fabrication of graphene nanoribbons. Nature 466(7305):470–473
Li X, Wang X, Zhang L, Lee S, Dai H (2008) Chemically derived, ultrasmooth graphene nanoribbon semiconductors. Science 319(5867):1229–1232
Berger C, Song Z, Li X, Wu X, Brown N, Naud C, Mayou D, Li T, Hass J, Marchenkov AN, Conrad EH, First PN, Heer WAD (2006) Electronic confinement and coherence in patterned epitaxial graphene. Science 312(5777):1191–1196
Hod O, Barone V, Scuseria GE (2008) Half-metallic graphene nanodots: a comprehensive first-principles theoretical study. Phys Rev B 77(3):035411
Deleuze MS, Huzak M, Hajgató B (2013) Half-metallicity of graphene nanoribbons and related systems: a new quantum mechanical El Dorado for nanotechnologies or a hype for materials scientists? J Mol Modeling 19(7):2699–2714
Huzak M, Deleuze MS, Hajgató B (2011) Half-metallicity and spin-contamination of the electronic ground state of graphene nanoribbons and related systems: an impossible compromise? J Chem Phys 135(10):104704
Schwabe T, Grimme S (2007) Double-hybrid density functionals with long-range dispersion corrections: higher accuracy and extended applicability. Phys Chem Chem Phys 9(26):3397–3406
Plasser F, Pašalić H, Gerzabek MH, Libisch F, Reiter R, Burgdōrfer J, Mūller T, Shepard R, Lischka H (2013) The multiradical character of one- and two-dimensional graphene nanoribbons. Angew Chem Int Ed 52(9):2581–2584
Torres AE, Guadarrama P, Fomine S (2014) Multiconfigurational character of the ground states of polycyclic aromatic hydrocarbons. A systematic study. J Mol Model 20(5):1–9
Muhich CL, Westcott JY, Morris TC, Weimer AW, Musgrave CB (2013) The effect of N and B doping on graphene and the adsorption and migration behavior of Pt atoms. J Phys Chem C 117(20):10523–10535
Rani P, Jindal VK (2012) Designing band gap of graphene by B and N dopant atoms. RSC Adv 3(3):802–812
Groves M, Chan A, Malardier-Jugroot C, Jugroot M (2009) Improving platinum catalyst binding energy to graphene through nitrogen doping. Chem Phys Lett 481(46):214–219
Wu M, Cao C, Jiang JZ (2010) Light non-metallic atom (B, N, O and F)-doped graphene: a first-principles study. Nanotechnology 21(50):505202
Miao L, Jia R, Wang Y, Kong CP, Wang J, Eglitis RI, Zhang HX (2017) Certain doping concentrations caused half-metallic graphene. J Saudi Chem Soc 21(1):111–117
Pekz R, Erkoç Ş (2009) A theoretical study of chemical doping and width effect on zigzag graphene nanoribbons. Phys E 42(2):110–115
Huang SF, Terakura K, Ozaki T, Ikeda T, Boero M, Oshima M, Ozaki T, Miyata S (2009) First-principles calculation of the electronic properties of graphene clusters doped with nitrogen and boron: analysis of catalytic activity for the oxygen reduction reaction. Phys Rev B 80(23):235410
Sharma S, Verma A (2013) A theoretical study of H2S adsorption on graphene doped with B, Al and Ga. Phys B 427:12–16
Zhang Hp, Luo Xg, Lin Xy, Lu X, Leng Y, Song Ht (2013) Density functional theory calculations on the adsorption of formaldehyde and other harmful gases on pure, Ti-doped, or N-doped graphene sheets. Appl Surf Sci 283:559–565
Zhang Hp, Luo Xg, Lin Xy, Lu X, Leng Y (2013) Density functional theory calculations of hydrogen adsorption on Ti-, Zn-, Zr-, Al-, and N-doped and intrinsic graphene sheets. Int J Hydrogen Energy 38(33):14269–14275
Al-Aqtash N, Al-Tarawneh KM, Tawalbeh T, Vasiliev I (2012) Ab initio study of the interactions between boron and nitrogen dopants in graphene. J Appl Phys 112(3):034304
Geldof D, Krishtal A, Blockhuys F, Van Alsenoy C (2011) An extension of the hirshfeld method to open shell systems using fractional occupations. J Chem Theory Comput 7(5):1328–1335
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr, JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2013) Gaussian 09 Revision D.01 (2013). Gaussian Inc., Wallingford CT
Bacskay GB (1981) A quadratically convergent Hartree–Fock (QC-SCF) method. Application to closed shell systems. Chem Phys 61(3):385–404
Bultinck P, Van Alsenoy C, Ayers PW, Carbó-Dorca R (2007) Critical analysis and extension of the hirshfeld atoms in molecules. J Chem Phys 126(14):144111–144111–9
Noodleman L (1981) Valence bond description of antiferromagnetic coupling in transition metal dimers. J Chem Phys 74(10):5737–5743
Noodleman L, Baerends EJ (1984) Electronic structure, magnetic properties, ESR, and optical spectra for 2-iron ferredoxin models by LCAO-X.alpha. valence bond theory. J Am Chem Soc 106(8):2316–2327
Noodleman L, Peng CY, Case DA, Mouesca JM (1995) Orbital interactions, electron delocalization and spin coupling in iron-sulfur clusters. Coord Chem Rev 144:199–244
Peverati R, Truhlar DG (2012) An improved and broadly accurate local approximation to the exchangecorrelation density functional: the MN12-L functional for electronic structure calculations in chemistry and physics. Phys Chem Chem Phys 14(38):13171–13174
Zhao Y, Truhlar DG (2006) A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J Chem Phys 125(19):194101
Čársky P, Hubač I (1991) Restricted Hartree-Fock and unrestricted Hartree-Fock as reference states in many-body perturbation theory: a critical comparison of the two approaches. Theor Chim Acta 80(4–5):407–425
Hirshfeld FL (1977) Bonded-atom fragments for describing molecular charge densities. Theor Chim Acta 44(2):129–138
Maroulis G (1995) Evaluating the performance of correlated methods in molecular property calculations: pattern recognition and clustering in spaces of theoretical descriptions. Int J Quantum Chem 55(2):173–180
Ferro-Costas D, Otero N, Graña A, Mosquera R (2012) A QTAIM-based energy partitioning for understanding the physical origin of conformational preferences: Application to the Z effect in O=C-X-R and related units. J Comput Chem 33(32):2533–2543
Otero N, Estévez L, Mandado M, Mosquera R (2012) An electron-density-based study on the ionic reactivity of 1,3-Azoles. Eur J Org Chem 12:2403–2413
Otero N, Mandado M, Mosquera R (2007) Nucleophilicity of indole derivatives: activating and deactivating effects based on proton affinities and electron density properties. J Phys Chem A 111(25):5557–5562
Otero N, González Moa M, Mandado M, Mosquera R (2006) QTAIM study of the protonation of indole. Chem Phys Lett 428(4–6):249–254
Janesko BG, Henderson TM, Scuseria GE (2009) Long-range-corrected hybrid density functionals including random phase approximation correlation: application to noncovalent interactions. J Chem Phys 131(3):034110
Chai JD, Head-Gordon M (2008) Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys Chem Chem Phys 10:6615–6620
Acknowledgements
This investigation is a part of the PICS Project (No-6115). N.O. thanks Xunta de Galicia for a grant under the I2C program and CNRS for 2-year postdoctoral contract. This work was granted access to the HPC resources of [CCRT/CINES/IDRIS] under the allocation 2014–2016 [Nos. i2014087031 and i2015087031] made by GENCI (Grand Equipement National de Calcul Intensif). We also acknowledge the “Direction du Numérique” of the “Université de Pau et des Pays de l’Adour” for the computing facilities provided.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published as part of the special collection of articles “In Memoriam of Claudio Zicovich.”
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Otero, N., Karamanis, P. & Pouchan, C. Hirshfeld-based atomic population analysis of the B, N doping effect in zigzag graphene nanoribbons: \(\pi\) electron density as requirement to follow the B, N doping guidelines. Theor Chem Acc 137, 16 (2018). https://doi.org/10.1007/s00214-017-2189-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-017-2189-5