
Abstract
The main challenge for the “hit-to-lead” stage in the drug discovery process relies on the accuracy of existing docking methods. In fact, accuracy of docking methods depends not only on the scoring function used to rank the poses but also on the ability of the docking method to reproduce the experimental binding mode. At this purpose, the performance of different approximations to properly dock and score compounds with known activity in a narrow range of IC50 values was analyzed. A set of five ATP-competitive CDK6 inhibitors and three receptor conformations for CDK6 were considered for analysis, and three methodologies were used and analyzed in order to include different degrees of receptor flexibility. Thus, a completely rigid receptor is considered when using Glide, while the so-called Induced Fit Docking Protocol accounts for receptor sidechain rearrangements. Finally, force field calculations were also performed in order to consider a completely flexible receptor.







Similar content being viewed by others
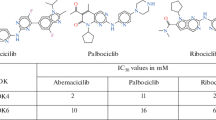
References
Malumbres M, Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9(3):153–166
Galons H, Oumata N, Meijer L (2010) Cyclin-dependent kinase inhibitors: a survey of recent patent literature. Expert Opin Ther Pat 20(3):377–404
Massague J (2004) G1 cell-cycle control and cancer. Nature 432(7015):298–306
Toogood PL et al (2005) Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6. J Med Chem 48(7):2388–2406
Lapenna S, Giordano A (2009) Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov 8(7):547–566
Malumbres M et al (2008) CDK inhibitors in cancer therapy: what is next? Trends Pharmacol Sci 29(1):16–21
Leach AR, Shoichet BK, Peishoff CE (2006) Prediction of protein-ligand interactions. Docking and scoring: successes and gaps. J Med Chem 49(20):5851–5855
Cheng T et al (2009) Comparative assessment of scoring functions on a diverse test set. J Chem Inf Model 49(4):1079–1093
Duca JS, Madison VS, Voigt JH (2008) Cross-docking of inhibitors into CDK2 structures. 1. J Chem Inf Model 48(3):659–668
Voigt JH et al (2008) Cross-docking of inhibitors into CDK2 structures 2. J Chem Inf Model 48(3):669–678
Schrödinger (2009) Glide, version 5.5. New York
Schrödinger (2010) Maestro, version 9.1. New York
Schrödinger (2009) Induced Fit Docking protocol (Schrödinger Suite 2009). New York, NY. p. Schrödinger Suite 2009: Glide version 5.5, Schrödinger, LLC, New York, NY, 2009; Prime version 2.1, Schrödinger, LLC, New York, NY, 2009
Kollman PA et al (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33(12):889–897
Lu H et al (2005) Crystal structure of a human cyclin-dependent kinase 6 complex with a flavonol inhibitor, fisetin. J Med Chem 48(3):737–743
Lu H, Schulze-Gahmen U (2006) Toward understanding the structural basis of cyclin-dependent kinase 6 specific inhibition. J Med Chem 49(13):3826–3831
Beattie JF et al (2003) Cyclin-dependent kinase 4 inhibitors as a treatment for cancer. Part 1: Identification and optimisation of substituted 4, 6-bis anilino pyrimidines. Bioorg Med Chem Lett 13(18):2955–2960
Ikuta M et al (2001) Crystallographic approach to identification of cyclin-dependent kinase 4 (CDK4)-specific inhibitors by using CDK4 Mimic CDK2 protein. J Biol Chem 276(29):27548–27554
Schulze-Gahmen U, Jung JU, Kim SH (1999) Crystal structure of a viral cyclin, a positive regulator of cyclin-dependent kinase 6. Structure 7(3):245–254
Jeffrey PD, Tong L, Pavletich NP (2000) Structural basis of inhibition of CDK-cyclin complexes by INK4 inhibitors. Genes Dev 14(24):3115–3125
Case DA et al (2005) The Amber biomolecular simulation programs. J Comput Chem 26(16):1668–1688
Viktor H et al (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct Funct Bioinform 65(3):712–725
Junmei W et al (2004) Development and testing of a general amber force field. J Comput Chem 25(9):1157–1174
William LJ et al (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79(2):926–935
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N · log(N) method for Ewald sums in large systems. J Chem Phys 98(12):10089–10092
Schrödinger (2009) LigPrep, version 2.3. New York
Friesner RA et al (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49(21):6177–6196
Sherman W et al (2006) Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 49(2):534–553
Schrödinger (2009) Glide 5.5 User Manual. Schrödinger Press
Hussain A, Melville JL, Hirst JD (2010) Molecular docking and QSAR of aplyronine A and analogues: potent inhibitors of actin. J Comput Aided Mol Des 24(1):1–15
Rodriguez A et al (2005) Assessment of the performance of cluster analysis grouping using pharmacophores as molecular descriptors. J Mol Struct Theochem 727(1–3):81–87
Berendsen HJC et al (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81(8):3684–3690
Ryckaert J-P, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23(3):327–341
Onufriev A, Bashford D, Case DA (2000) Modification of the generalized born model suitable for macromolecules. J Phys Chem B 104(15):3712–3720
Jörg W, Peter SS, Still WC (1999) Optimization of Gaussian surface calculations and extension to solvent-accessible surface areas. J Comput Chem 20(7):688–703
Onufriev A, Bashford D, Case DA (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins Struct Funct Bioinform 55(2):383–394
Sadiq SK et al (2010) Accurate ensemble molecular dynamics binding free energy ranking of multidrug-resistant HIV-1 proteases. J Chem Inf Model 50(5):890–905
Zhang X, Li X, Wang R (2009) Interpretation of the binding affinities of PTP1B inhibitors with the MM-GB/SA method and the X-score scoring function. J Chem Inf Model 49(4):1033–1048
Acknowledgments
The Spanish Ministry of Science and Technology supported this work through project CTQ2006-06588/BQU. This work was also supported in part by the Generalitat de Catalunya through project 2009SGR1308. We are also grateful to the Departament d’Universitat, Recerca i Societat de la informació de la Generalitat de Catalunya i del Fons Social Europeu.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published as part of the special issue celebrating theoretical and computational chemistry in Spain.
Electronic supplementary material
Table S-1 and S-2 containing detailed information about docking protocols. Table S-3 with the mean values of the three complexes for each MMPB/GBSA term and partial summation.
Rights and permissions
About this article
Cite this article
Delgado-Soler, L., Ariñez-Soriano, J., Granadino-Roldán, J.M. et al. Predicting binding energies of CDK6 inhibitors in the hit-to-lead process. Theor Chem Acc 128, 807–823 (2011). https://doi.org/10.1007/s00214-010-0857-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00214-010-0857-9