
Abstract
Next Generation Sequencing (NGS) is providing new methodologies to improve and complement traditional genetic approaches. These strategies, collectively termed NGS-enabled genetics, consist of identifying variation in bulks of plants that have been assembled based on a specific phenotype of interest. We examined NGS-enabled genetics in hexaploid wheat by using near isogenic lines (NIL) differing across a specific disease resistance locus. RNA-Seq of NILs allowed the identification of SNPs across this locus and helped distinguish allelic SNPs from homoeologous variants. F2 bulks were assembled based on opposing disease resistance phenotypes and the frequency of the informative allelic SNPs was examined across bulks using RNA-Seq. Variants enriched in the corresponding bulks are expected to be most closely linked to the phenotype of interest and were prioritized for validation. Recent advances in cereal genomics in the form of wheat gene models, sequenced diploid progenitors, and the advances in the Chromosome-based Survey Sequencing Project enabled us to develop a pipeline to automatically design SNP-based markers. These high-throughput assays were used to genotype the original individuals used to assemble the bulks and to generate a genetic map across the target locus. Linked markers are now being incorporated into marker assisted selection programs by breeders.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others
Keywords
- Bulk frequency ratio
- Bulked segregant analysis
- Genotyping
- KASP
- Marker assisted selection
- Near isogenic lines
- Next generation sequencing
- RNA-Seq
Introduction
Wheat is among the most important crops in the world providing over 20 % of the world’s calorie and protein intake (FAO 2012). Genetic improvement and breeding, however, are hampered by the large proportion of repetitive elements (>80 %, Flavell et al. 1974) across its large genomes and the polyploid nature of hexaploid wheat which often leads to functional redundancy among homoeologous genes (Avni et al. 2014). The advances in next generation sequencing (NGS) technologies are creating new opportunities to exploit the variation in the wheat genome for crop improvement, both in repetitive regions (insertion site-based polymorphism (ISBP) markers, Paux et al. 2010) and in low copy regions (gene based single nucleotide polymorphisms (SNPs), Allen et al. 2013).
The challenges that are thus emerging are how to prioritize the large number of SNPs available both in public databases, for example Cerealsdb.uk.net (Wilkinson et al. 2012), and those obtained with Next Generation Sequencing (NGS), as well as how to convert these into high-throughput (HTP) markers for breeding. One option is to exploit synteny between grass species (Moore et al. 1995), although this approach is limited to gene-based SNPs. The recent release of the Wheat Chromosome-based Survey Sequencing Project coordinated by the International Wheat Genome Sequencing Consortium (IWGSC) allows broad scale localisation to a particular homoeologous chromosome arm, although resolution is limited.
Wheat Genetics
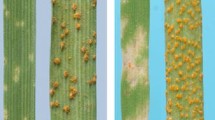
A complementary approach is to exploit genetics to decrease the background complexity of the wheat genome. Near isogenic lines (NILs) that only differ across a specific target region can be a powerful resource for this purpose. A great example is the set of Avocet-S NILs for major disease resistance (R) genes against yellow rust (Yr) developed by Colin Wellings and colleagues at the University of Sydney. We have used the Avocet S-Yr15 NILs (Fig. 22.1a) to develop a segregating BC7F2 population that was phenotyped through virulence assays to the wheat yellow rust pathogen (Puccinia striiformis). Resistant plants, composed of both homozygous and heterozygous individuals across the Yr15 locus, were pooled into a resistant bulk. In an analogous manner, susceptible plants were grouped into a susceptible bulk (Fig. 22.1b). This strategy, first published as bulked segregant analysis (BSA) in 1991 (Michelmore et al. 1991), can now be further exploited with NGS approaches. An initial complexity reduction is achieved with the Avocet NILs whose main difference is across the R gene locus. This list of SNPs can then be further refined by the BSA approach. In this study we highlight how the combination of genetic and genomic resources, along with methods such as BSA, can be used to leverage new biological research and develop HTP markers for breeding.

Segregating population for Yr15. (a) Resistant and susceptible phenotypes of Avocet S and Avocet S + Yr15 challenged with the yellow rust pathogen (Puccinia striiformis), respectively. Scale bar; 1 cm. (b) Segregating lines were developed by crossing a homozygous resistant NIL, Avocet S + Yr15, to Avocet S. Heterozygous plants were then self-pollinated to produce a segregating F2 population with an expected 3:1 segregation ratio for resistant and susceptible individuals, respectively. The resistant and susceptible plants were grouped by phenotype into the corresponding bulks. The resistant bulk includes both homozygous and heterozygous individuals across the Yr15 locus as exemplified by the red squares on the wheat chromosomes
Wheat Genomics
A high quality reference genome of bread wheat hasn’t yet been completed. In the past few years, however, the wheat community has produced valuable genomic resources that can be used as a proxy reference: a whole genome shotgun (WGS) assembly of Chinese Spring based on 454 technology (Brenchley et al. 2012); the reference genomes for the diploid progenitor species Triticum uratu (2n = 14;AA) (Ling et al. 2013) and Aegilops tauschii (2n = 14;DD) (Jia et al. 2013); an assembly of the close Triticeae relative Hordeum vulgare L. (2n = 14;HH) (International Barley Genome Sequencing et al. 2012); and the genomic contigs from the Chromosome-based Survey Sequencing (CSS; International Wheat Genome Sequencing Consortium 2014). We can use the listed collections as supporting information, bearing in mind that discrepancies are expected for the related organisms and that specific genes being studied may not be present in Chinese Spring. Nevertheless, using the available genomic sequences facilitates NGS analyses that depend on a reliable genome reference.
SNPs that can be used to map genes of interest are becoming increasingly available. The CerealsDB website holds ~100,000 SNPs from British varieties (Allen et al. 2013) obtained by transcriptome and exome sequencing, although only a fraction (<8 %) have been converted into functional HTP assays. Recently a subset of these have been incorporated into the ~82,000 iSelect array coordinated by Eduard Akhunov at Kansas State University (Wang et al. 2014) which follows the 9,000 SNP array (Cavanagh et al. 2013). These SNP arrays are providing an extremely valuable resource for the community in diversity and association studies. Their use however is limited for large mapping populations because of their cost, the difficulty in interpreting data when using diploid or alien introgressions, and the difficulty to reliably call heterozygous individuals. Because of these reasons, alternative approaches are sometimes required to identify polymorphisms across target regions.
NGS is providing different methodologies to identify putative variations. WGS sequencing consists on sequencing random fragments of genomic DNA without any selection except for the fragment size. The reads are then aligned to a reference and SNPs can be called from the alignments. In several model organisms, these re-sequencing NGS approaches have been combined with bulks/pools of phenotypically distinct individuals to identify SNPs that are closely linked with the gene of interest within a single experiment.
These approaches, collectively termed as NGS-enabled genetics (Schneeberger and Weigel 2011), are rapidly evolving and have used different strategies. MutMap produces segregant populations from seeds with induced mutations and then bulks are sequenced using WGS (Abe et al. 2012; Takagi et al. 2013b). QTL-seq takes two plants with opposite phenotypes and the progeny with extreme phenotypes are bulked and their DNA is extracted for sequencing (Takagi et al. 2013a). Renseq, focuses on R-genes by designing baits from known resistance genes and performing targeted resequencing (Jupe et al. 2013). The mentioned techniques do not scale well in wheat because they rely on having a reliable reference sequence and relatively small genome size. A full Illumina HiSeq 2500 can produce 600-Gbp in a single run, providing 35-fold coverage of the complete wheat genome. However, this translates into less than two reads for each position per individual for a bulk of 20 plants.
To reduce the complexity of the data, we propose to focus on sequencing the wheat transcriptome using RNA-Seq (Westermann et al. 2012) instead of genomic DNA. By sequencing the transcribed RNA we can use short read aligners and use transcript assemblies as reference (Fig. 22.2a). At first we used the wheat UniGenes set available NCBI, which consist of collapsed homoeologous transcripts from a myriad of sources (Pontius et al. 2002). More recently, we have shifted to a phased transcriptome which has gene models separated by the corresponding genome (Krasileva et al. 2013).

RNA-Seq NGS-enabled genetics. (a) Representation of a typical RNA-Seq output; the method excludes non-coding regions from the genome and the coverage is correlated to the expression of the gene. (b) Illustration of a non-informative homoeologous SNP (G181T) present in both parental lines, and an informative allelic SNP (G184A), only present in the resistant progenitor Avocet S + Yr15. The consensus sequences from the parental genotypes include this information in the form of ambiguity codes (K and R, respectively). In the bulks, the individual reads align across theFig. 22.2 (continued) reference sequence, with matches indicated by dots, and polymorphisms at positions 181 and 184 indicated by the corresponding nucleotide variants at those positions. The SNP index is calculated as the frequency of the informative allelic SNP in each bulk. The Bulk Frequency Ratio is the quotient of the resistant and susceptible bulk SNP Indexes. (c) Primer design: The allelic SNP (G184A) is used at the 3′ end of the differentiating primers. For the common primer, a homoeologous SNP is selected for the 3′ end to make the marker genome-specific. (d) The KASP assay output shows the intensities of the HEX and FAM fluorescence of individual plants as a single dot. The clusters near the X and Y-axis are composed of homozygous individuals, while the central cluster contains heterozygous plants
RNA-Seq captures the full range of the dynamic spectrum of the transcriptome, an advantage when compared to array platforms that are restricted to the pre-defined set of variants incorporated into the array design. SNPs can be identified either by aligning to a known transcriptome or by de novo assembly over the transcriptome (Grabherr et al. 2011). With the use of F2 populations it is possible to create a panel of putative SNPs that enables haplotype analysis without a priori knowledge of the positions of the loci (Trick et al. 2012). RNA-Seq allows rapid access to SNPs in wheat and it scales well as the transcriptome is several orders of magnitude smaller than the genome, ~80 Mb compared to 17 Gb, respectively. In principle, it is possible to sequence the transcriptome with an average coverage over 900x (keeping in mind variations of coverage due to expression) in a single HiSeq 2,500 lane, as opposed to just over 1.5x of genomic sequence.
The original purpose of RNA-Seq is to characterize expression levels of genes, which can bias the SNP calling, as the assumption of uniform coverage is not valid. To overcome these biases and analyse the volume of data produced by NGS, bioinformatics pipelines and access to high performance computing are required. Although this is a potential barrier for adoption, new web-based user-friendly graphical interfaces, such as Galaxy (Goecks et al. 2010), are empowering new users to access high-performance computing facilities. For NGS-enabled genetics in wheat, we propose a pipeline that integrates BSA (Michelmore et al. 1991; Trick et al. 2012), syntenic information from related grasses, and the use of the CSS to aid in the design of genome specific primers from putative SNPs.
SNP Selection and Marker Design
To identify loci linked to a trait we use BSA and extend it by using RNA-Seq instead of targeting known markers. We sequence the parental genotypes (Avocet S and Avocet S + Yr15, Fig. 22.1a, b) and generate a consensus reference by aligning the reads to the UniGenes and gene models described above (Krasileva et al. 2013). Although genome-specific references are used when possible, there are still cases where multiple homoeologues will align to a common reference (as illustrated in Fig. 22.2b). These homoeologous variants (exemplified by the G > T variant at position 181; K in consensus) will generate ambiguity codes within both parental consensus sequences and can therefore be excluded. Real allelic SNPs between the parental genotypes (exemplified by the G > A variant at position 184; R in consensus) are distinguished by the presence in one, but not the other parental consensus sequence (Fig. 22.2b).
These allelic SNPs are then examined further in the alignments of the RNA-Seq reads from the susceptible and resistant bulks. To identify enriched and depleted SNPs we first calculate the SNP Index, which is the proportion of the non-consensus base at the positions previously identified (A at position 184 according to the example in Fig. 22.2b; Takagi et al. 2013a). Then, we calculate the bulk frequency ratios (BFR), which is the ratio between the SNP Indexes of the resistant and susceptible bulks (Trick et al. 2012). The BFR helps reduce the noise generated by differential expression of homoeologous genes and accounts for the presence of alternative bases at any given position. A closely linked SNP to the R-gene should generate a very high BFR since the resistant bulk will carry exclusively plants with the resistant allele, whereas the susceptible bulk should be devoid of any plants carrying the resistant allele. As one moves further away from the R-gene, recombination events occur between the gene and the candidate SNPs decreasing the BFR.
Once a list of SNPs with the corresponding BFRs is obtained, these can be prioritized in several ways before independent validation as genetic markers. First and foremost is the BFR value itself; the higher the BFR the most likely the SNP is genetically linked to the gene. Second, we can align the candidate genes to the genome of syntenic species such as Hordeum vulgare, Brachypodium distachyon and Oryza sativa to identify the orthologous genes and start to identify syntenic regions with high BFRs. More recently, we have started to align the genes to the CSS assemblies to locate the chromosome arms with highest BFR. The putative SNPs with enriched BFR can then be converted into HTP SNP assays (see next section) and genotyped across the individuals that were used to assemble the bulks. This generates a genetic map with markers across the R-gene locus. If the interval is syntenic to one of the sequenced grass genomes, an additional round of SNP selection can be performed based on synteny and using slightly more relaxed parameters to establish the BFR cut-off.
High-Throughput Genotyping
A key issue is to move from in silico SNPs into a HTP SNP assay that can be amenable to both researchers and wider applications such as marker assisted selection (MAS) in breeding programs. In this regard, it is particularly important to have genome specific SNP assays that allow the screening of germplasm where heterozygotes need to be easily identified. The IWGSC CSS assemblies facilitate this objective by allowing us to generate a multiple alignment of the reference sequence containing the SNP of interest with the assemblies from the three homoeologous genomes (Fig. 22.2c). In this manner, homoeologous SNPs can be readily identified and incorporated into the primer design to assay the SNP only in the genome of interest. This is particularly applicable to end point fluorometric assays, such as KASP (Allen et al. 2013), that identify SNPs using two differentially labeled primers with the SNP at the 3′ end of the primers (boxed sequences with SNP in bold; Fig. 22.2c). A common reverse primer can be designed across a homoeologous SNP to generate the genome specific amplification. This results in a HTP assay that can be readily used to genotype heterozygous individuals (Fig. 22.2d). In the past, the generation of genome-specific assays has been a time consuming task (Chao et al. 2008) but with the use of this pipeline the creation of allele specific SNPs has been simplified. We developed a web-based user-friendly interface (PolyMarker, Ramirez-Gonzalez et al. 2015a) to make this pipeline readily available to the community (http://polymarker.tgac.ac.uk).
Final Remarks
Despite the complexity of the wheat genome and the draft status of the genomic reference, it is possible to advance in the development of NGS-enabled genetic approaches in hexaploid wheat. RNA-Seq of NILs facilitates the identification of SNPs and helps distinguish these as informative allelic SNPs or non-informative homoeologous variants. RNA-Seq of NIL-derived F2 bulks helps identify those SNPs that are most closely linked to the phenotype of interest. With the aid of the recently released CSS, putative SNPs can be rapidly converted into HTP assays that can be incorporated into MAS improvement programs by breeders (Ramirez-Gonzalez et al. 2015b). These approaches will continue to improve in resolution as physical maps improve in wheat and homoeologous relationships between transcripts are more precisely defined.
References
Abe A, Kosugi S, Yoshida K et al (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30:174–178
Allen AM, Barker GL, Wilkinson P et al (2013) Discovery and development of exome-based, co-dominant single nucleotide polymorphism markers in hexaploid wheat (Triticum aestivum L.). Plant Biotechnol J 11:279–295
Avni R, Zhao R, Pearce S et al (2014) Functional characterization of GPC-1 genes in hexaploid wheat. Planta 239:313–324
Brenchley R, Spannagl M, Pfeifer M et al (2012) Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491:705–710
Cavanagh CR, Chao S, Wang S et al (2013) Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc Natl Acad Sci U S A 110:8057–8062
Chao S, Zhang W, Akhunov E et al (2008) Analysis of gene-derived SNP marker polymorphism in US wheat (Triticum aestivum L.) cultivars. Mol Breed 23:23–33
FAO (2012) FAOSTAT. http://faostat.fao.org/site/609/DesktopDefault.aspx?PageID=609. Accessed 5 Dec 2013
Flavell RB, Bennett MD, Smith JB, Smith DB (1974) Genome size and the proportion of repeated nucleotide sequence DNA in plants. Biochem Genet 12:257–269
Goecks J, Nekrutenko A, Taylor J (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11:R86–R86
Grabherr MG, Haas BJ, Yassour M et al (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
International Wheat Genome Sequencing Consortium (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345(6194):1251788
International Barley Genome Sequencing Consortium, Mayer KF, Waugh R et al (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491:711–716
Jia J, Zhao S, Kong X et al (2013) Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496:91–95
Jupe F, Witek K, Verweij W et al (2013) Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J 76:530–544
Krasileva KV, Buffalo V, Bailey P et al (2013) Separating homeologs by phasing in the tetraploid wheat transcriptome. Genome Biol 14:R66–R666
Ling H-Q, Zhao S, Liu D et al (2013) Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496:87–90
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci U S A 88:9828–9832
Moore G, Devos KM, Wang Z, Gale MD (1995) Cereal genome evolution: grasses, line up and form a circle. Curr Biol 5:737–739
Paux E, Faure S, Choulet F et al (2010) Insertion site-based polymorphism markers open new perspectives for genome saturation and marker-assisted selection in wheat. Plant Biotechnol J 8:196–210
Pontius JU, Wagner L, Schuler GD (2002) UniGene: a unified view of the transcriptome chapter 21. In: McEntyre J, Ostell J (eds) The NCBI handbook [Internet]. National Center for Biotechnology Information (US), Bethesda
Ramirez-Gonzalez RH, Uauy C, Caccamo M (2015a) PolyMarker: a fast polyploid primer design pipeline. Bioinformatics. doi:10.1093/bioinformatics/btv069
Ramirez-Gonzalez RH, Segovia V, Bird N et al (2015b) RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnol J 13:613–624
Schneeberger K, Weigel D (2011) Fast-forward genetics enabled by new sequencing technologies. Trends Plant Sci 16:282–288
Takagi H, Abe A, Yoshida K et al (2013a) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74:174–183
Takagi H, Uemura A, Yaegashi H et al (2013b) MutMap-Gap: whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol 200:276–283
Trick M, Adamski NM, Mugford SG et al (2012) Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. BMC Plant Biol 12:14
Wang S, Wong D, Forrest K et al (2014) Characterization of polyploid wheat genomic diversity using a high-density 90 000 single nucleotide polymorphism array. Plant Biotechnol J 12:787–797
Westermann AJ, Sa G, Vogel J (2012) Dual RNA-seq of pathogen and host. Nat Rev Microbiol 10:618–630
Wilkinson PA, Winfield MO, Barker GLA et al (2012) CerealsDB 2.0: an integrated resource for plant breeders and scientists. BMC Bioinformatics 13:219–219
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this paper
Cite this paper
Ramirez-Gonzalez, R.H., Segovia, V., Bird, N., Caccamo, M., Uauy, C. (2015). Next Generation Sequencing Enabled Genetics in Hexaploid Wheat. In: Ogihara, Y., Takumi, S., Handa, H. (eds) Advances in Wheat Genetics: From Genome to Field. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55675-6_22
Download citation
DOI: https://doi.org/10.1007/978-4-431-55675-6_22
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55674-9
Online ISBN: 978-4-431-55675-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)