
Abstract
Correct track-to-track association is crucial to the construction of cell lineage trees as well as the discovery of novel biological phenomenon that occur at rare frequencies. In this paper, an ant colony optimization based heuristic approach is proposed to link potential tracks through minimizing the cost function that mainly occurs on the fragmented intervals with the constraint of maximum inter-frame displacement. Specifically, both cell motion and morphology are emphasized in the defined cost function, and two decisions are made respectively to recover the mitotic and non-mitotic cases. Our method has proven to be feasible that can repair the broken tracklets caused by large migration, occlusion and mitosis missing, as well as false positives and missed detections, and can effectively help the construction of reliable cell lineage trees.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
The analysis of cellular behaviors is crucial for human disease diagnose and biomedical research. The existing automated cell tracking methods mainly focus on frame-by-frame association that can achieve high tracking accuracy in continuous image sequences [1, 2], but it still exists some problems due to noise or segmentation error. For example, when a false negative segmentation occurs among a neighboring cell cluster, the normal tracking methods may cause a track missing. To resolve the problem, many global spatio-temporal data association approaches have been proposed to associate multiple trajectories over time of which the most well-known are the Multi-Hypothesis Tracking (MHT) [3] and Joint Probabilistic Data Association Filters (JPDAF) [4]. In [5], a batch algorithm, which utilizes information from all images in the image sequence, incorporates mitosis, apoptosis and other events into the same probabilistic framework for track linking. To reduce the computational cost, Huang et al. [6] first generate reliable track fragments and then stitch them by Hungarian algorithm.
In this paper, we introduce an ant colony optimization [7] based on heuristic approach linking potential tracks through minimizing the cost function that mainly occurs on the fragmented intervals with the constraint of maximum inter-frame displacement. In our work, each track segment is assumed to be associated with an ant colony. All ants in the colony start their foraging behaviors from the initial position of the track, and each ant makes decision via both the heuristic function and the pheromone intensity on the track. During decision, two events, namely, migration without mitosis and migration with mitosis, are considered to link the potential tracks with identical ancestry. As iterations increase, the trail pheromone field is continuously strengthened, and the trails with high pheromone intensity above a prespecified threshold are finally extracted. The rest of this paper is organized as follow: Sect. 2 details our ant-inspired track-to-track recovery method. We provide our experimental results in Sect. 3 and conclude this paper in Sect. 4.
2 Methods
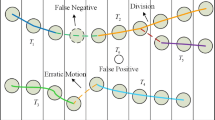
Figure 1(a) illustrates an example of a set of broken cell tracks produced by an automated tracking algorithm, where cell occlusion, erratic motion and division probably cause the undetected candidates, while the presence of clutter usually generates a set of short track candidates. In order to construct correct cell lineage trees, a track recovery approach is needed to link the associated broken tracks and remove the false alarms, as shown in Fig. 1(b).

Examples of broken tracks and overall trajectories
Let \(p_{u}^{k}\) and \(p_{v}^{k+d}\), respectively, denote the states of cell u in frame k and cell v in frame \(k+d\), where d is a positive integer with the constraint of \(d \le 3\). In our work, a very small value is assigned by ant decision if an ant travels on the existing tracks while we formulate a new cost function between the cell \(p_{u}^{k}\) and the cell \(p_{v}^{k+d}\) that only occurs on the fragmented tracks as
where \(\alpha _{1}\) is a constant range in [0, 1], \(E_{dis}(p_{u}^{k},p_{v}^{k+d})\) and \(E_{area}(p_{u}^{k},p_{v}^{k+d})\), respectively, represent costs of distance and overlap of area between cells \(p_{u}^{k}\) and \(p_{v}^{k+d}\). The cost \(E_{dis}\) is defined as
where H and W denote the height and width of an image respectively. The cost \(E_{area}\) is formulated by
where \(A(p_{u}^{k})\) refers to the area of the cell \(p_{u}^{k}\), \(A(p_{v}^{k+d})\) refers to the area of the cell \(p_{v}^{k+d}\). We observe that the closer the two cells, the smaller the cost function.
An one-one mapping is defined between a track and an ant colony. With the known cell states of each track, we put a group of ant colony starting foraging from the initial position of a tracklet. Then each ant makes decision to move to the next cell in sequence with constraint of maximum inter-frame displacement.
Firstly, each ant a at the current position \(p_{u}^{k}\) finds the set of candidates \(N_{a}^{k+d}\) to be visited in frame \(k+d\). If \(|N_{a}^{k+d}| \ge 2\), the detection of mitotic event is carried out. Let \(p_{m}^{k+d}\) and \(p_{n}^{k+d}\) denote the states of any two cell candidates, namely cell m and cell n, in frame \(k+d\), the similarity score \(S(p_{u}^{k},p_{m}^{k+d},p_{n}^{k+d})\) is defined as
where \(\beta _{i}\) is a constant range in [0, 1] and \(\sum \limits _{i=1}^{3}\beta _{i}=1\), \(S_{area}(\cdot )\), \(S_{dis}(\cdot )\) and \(S_{def}(\cdot )\), respectively, denote the similarity scores of area, distance and deformation of cells, which are defined as
where \(L_{h}(\cdot )\) and \(L_{w}(\cdot )\), respectively, denote the length of major and minor axes of the estimated cell contour.
If the similarity score \(S(p_{u}^{k},p_{m}^{k+d},p_{n}^{k+d})\) is above a prespecified threshold \(\varepsilon _{1}\), two ants will select \(p_{m}^{k+d}\) and \(p_{n}^{k+d}\) respectively and the selected cells are disjoint with \(p_{m}^{k+d}\cap p_{n}^{k+d}=\varnothing \). If no mitotic event is detected, that is, the similarity score \(S(p_{u}^{k},p_{m}^{k+d},p_{n}^{k+d})<\varepsilon _{1}\) or \(|N_{a}^{k+d}|=1\), each ant a at the current position \(p_{u}^{k}\) selects a candidate \(p_{v}^{k+d}\) according to
where \(\tau _{p_{u}^{k},p_{v}^{k+d}}\) denotes the amount of trail pheromone on path \((p_{u}^{k}, p_{v}^{k+d})\) and \(\eta _{p_{u}^{k},p_{v}^{k+d}}\) denotes the heuristic information with \(\eta _{p_{u}^{k},p_{v}^{k+1}}=1/E(p_{u}^{k},p_{v}^{k+d})\). In our method, we first set \(d=1\). If \(|N_{a}^{k+d}|=0\), we expand the ant’s step size as \(d \leftarrow d+1\), and then the ant repeats the above decision-making process.

Once ant a has made a decision, a local update process of trail pheromone is conducted on its visited path by
where \(\varDelta \tau _{c}\) denotes a fixed amount of pheromone.
When all ants in the colony complete their tours, an individual global trail pheromone update is defined as
where \(Q_{1}\) is a constant, \(E_{bt}\) denotes the cost of the best tour \(P_{bt}\).
Once all ant colonies complete their foraging, a global update trail pheromone on all ant colonies is performed by
where \(\sum \limits _{i=1}^{m}E_{bt}^{i}\) denotes total cost of the set of best tours \(\{P_{bt}^{i}\}_{i=1}^{m}\) of all ant colonies in current iteration, \(P_{bt}^{i}\) denotes the ith best tour of \(\{P_{bt}^{i}\}_{i=1}^{m}\).
As iterations increase, the trail pheromone field is continuously strengthened. Once reaching the maximum value of iteration, the trails with high pheromone intensity above a prespecified threshold \(\varepsilon _{2}\) are finally extracted by morphological operations. The main framework of our method is summarized in Algorithm 1.
3 Experimental Results
To test the performance of our proposed track recovery method, we implemented it in MATLAB(R2014a) on a 2.6 GHz processor computer with 16G random access memory. In this section, we use two image sequences from publicly available website at http://www.celltrackingchallenge.net/. The two cell image sequences include real N2DL-Hela+ dataset and pancreatic stem cell dataset. With the reliable track fragments results, our track recovery method proved to be effective. The parameters used in our approach mainly focus on ant colony setting, we have \(\alpha =3\), \(\beta =2\), and \(\rho =0.3\). The initial value of the pheromone field is \(c=0.0005\). We set the mitotic similarity threshold \(\varepsilon _{1}=0.85\), the pheromone extracting threshold \(\varepsilon _{2}=0.05\) and the maximum iteration parameter \(t_{max}=20\). We find that three factors of similarity score in (4), i.e., area, distance and deformation of cells, are crucial to the recovery of mitotic events, which affects the track accuracy. To observe the effect of these factors on similarity score, we compare the performance of different combinations using several measure metrics.
The track recovery results on two cell image sequences are, respectively, shown in Fig. 2 and Fig. 3, which illustrate the track recovery results including lineages and cell contours based on the raw fragmented tracks.

Track recovery results of N2DL-Hela+ dataset

Track recovery results of pancreatic stem cell dataset (Color figure online)
In N2DL-Hela+ dataset, there are 25 tracklets with different track colors before track recovery in Fig. 2(a), which is generated by cell erratic motion, cell division, and cell occlusion. After the proposed recovery method, the true reliable tracks have been stitched to be 19 entire trajectories as shown in Fig. 2(b). From the lineage trees as shown in Fig. 2(c), the track segments marked in red circles have been correctly linked into longer trajectories as illustrated in Fig. 2(d). In pancreatic stem cell dataset, the 35 tracks in Fig. 3(a) including false positives and false negatives have been recovered to be 25 entire tracks, and false positive tracks are removed as shown in Fig. 3(b). The comparison lineage trees are also shown in Fig. 3(c) and Fig. 3(d) with red circle marks.
Obviously, our track recovery method can fix broken tracks and restore mitotic events, and finally can obtain the whole reliable cell lineage trees. Figure 4 shows the resulting trail pheromone field. The color corresponds to pheromone intensity, and the trails with the amount of pheromone greater than a prespecified threshold are extracted.

2-D trail pheromone field
For performance evaluation, we use the complete tracks (CT) [8], which shows how good an approach is at reconstructing complete reference tracks and is defined as \(CT=\dfrac{2T_{rc}}{T_{gt}+T_{c}}\), where \(T_{rc}\) denotes the number of completely reconstructed reference tracks, \(T_{gt}\) denotes the number of all reference tracks and \(T_{c}\) denotes the number of all computed tracks. To jointly evaluate the errors of label and cardinality of tracks, the metric of optimal sub-pattern assignment metric for track (OSPA-T) [9] is introduced as well. We compare the performance of different combinations of three factors of similarity score in (4), i.e., area, distance and deformation of cells, as shown in Table 1. We observe that the performance is better when all three factors are taken into account simultaneously, in which the mitotic events are accurately detected.
4 Conclusion
In this paper, we proposed an ant-inspired track recovery approach linking potential tracks through minimizing the cost function that mainly occurs on the fragmented intervals with the constraint of maximum interframe displacement. Through constant iterations, the completed repaired trails were obtained through minimizing total cost. It is demonstrated that our proposed approach successfully obtain reliable cell trajectories and lineage trees by stitching sets of true track segments and reconstructing missing mitosis branching.
References
Padfield, D., Rittscher, J., Roysam, B.: Coupled minimum-cost flow cell tracking for high-throughput quantitative analysis. Med. Image Anal. 15(4), 650–668 (2011)
Benlian, X., Shi, J., Mingli, L., Cong, J., Wang, L., Nener, B.: An automated cell tracking approach with multi-bernoulli filtering and ant colony labor division. IEEE/ACM Trans. Comput. Biol. Bioinform. (2019). https://doi.org/10.1109/TCBB.2019.2954502
Reid, D.B.: An algorithm for tracking multiple targets. IEEE Trans. Autom. Control 24(6), 1202–1211 (1979)
Fortmann, T.E., Bar-Shalom, Y., Scheffe, M.: Sonar tracking of multiple targets using joint probabilistic data association. IEEE J. Oceanic Eng. 8(3), 173–184 (2003)
Magnusson, K.E.G., Jaldén, J., Gilbert, P.M., Blau, H.M.: Global linking of cell tracks using the Viterbi algorithm. IEEE Trans. Med. Imaging 34(4), 911–929 (2014)
Huang, C., Wu, B., Nevatia, R.: Robust object tracking by hierarchical association of detection responses. In: Forsyth, D., Torr, P., Zisserman, A. (eds.) ECCV 2008. LNCS, vol. 5303, pp. 788–801. Springer, Heidelberg (2008). https://doi.org/10.1007/978-3-540-88688-4_58
Dorigo, M., Gambardella, L.M.: Ant colony system: a cooperative learning approach to the traveling salesman problem. IEEE Trans. Evol. Comput. 1(1), 53–66 (1997)
Ulman, V., et al.: An objective comparison of cell-tracking algorithms. Nat. Methods 14(12), 1141 (2017)
Ristic, B., Vo, B.-N., Clark, D., Vo, B.-T.: A metric for performance evaluation of multi-target tracking algorithms. IEEE Trans. Signal Process. 59(7), 3452–3457 (2011)
Acknowledgement
This work was supported by National Natural Science Foundation of China (No. 61673075 and No. 61876024), 333 Project of Jiangsu Province (No. BRA2019284), and Project of talent peak of six industries (2017-DZXX-001).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this paper

Cite this paper
Wu, D., Bu, H., Xu, B., Lu, M., Sun, Z. (2020). An Ant-Inspired Track-to-Track Recovery Approach for Construction of Cell Lineage Trees. In: Tan, Y., Shi, Y., Tuba, M. (eds) Advances in Swarm Intelligence. ICSI 2020. Lecture Notes in Computer Science(), vol 12145. Springer, Cham. https://doi.org/10.1007/978-3-030-53956-6_20
Download citation
DOI: https://doi.org/10.1007/978-3-030-53956-6_20
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-53955-9
Online ISBN: 978-3-030-53956-6
eBook Packages: Computer ScienceComputer Science (R0)