
Abstract
Sortase is a cysteine-transpeptidase that anchors LPXTG-containing proteins on the Gram-positive bacterial cell wall. Previously, sortase was considered to be an important factor for bacterial pathogenesis and fitness, but not cell growth. However, the Actinomyces oris sortase is essential for cell viability, due to its coupling to a glycosylation pathway. In this chapter, we describe the methods to generate conditional srtA deletion mutants and identify srtA suppressors by Tn5 transposon mutagenesis. We also provide procedures for analyzing cell morphology of this mutant by thin-section electron microscopy. These techniques can be applied for analyses of other essential genes in A. oris.
1 Introduction
Actinomyces oris is a cariogenic bacterium that is important for the formation of oral biofilm, commonly known as dental plaque [1, 2]. The ability of A. oris to cultivate biofilm is dependent on the adhesive type 1 and 2 fimbriae, which, via a C-terminal cell-wall-sorting signal (CWSS), are assembled and anchored to the cell surface by cysteine transpeptidase sortase (Srt) enzymes. Type 1 fimbriae, composed of the fimbrial shaft FimP and tip fimbrillin FimQ, facilitate A. oris colonization by binding to proline-rich salivary deposits on the tooth surface [3]. Type 2 fimbriae, consisted of the fimbrial shaft FimA and tip fimbrillins FimB or CafA, promote biofilm development by mediating interactions with bacterial co-colonizers and host cells [4, 5]. Type 1 and 2 fimbriae are assembled by the pilus-specific sortases SrtC1 and SrtC2, respectively, but they are linked to the cell wall by a single housekeeping sortase (SrtA). In additional to targeting type 1 and 2 fimbriae, SrtA is also predicted to anchor 14 non-pilus proteins containing the CWSS [6].
Housekeeping sortases are conserved in Gram-positive bacteria and serve as major virulence factors. The first housekeeping sortase was discovered in Staphylococcus aureus by Schneewind’s group in 1999 [7]. Since then, many others have been characterized, but none were found to be required for viability [8, 9]. Recently, Wu and colleagues revealed that the A. oris housekeeping sortase (SrtA) is an exception [6]. Multiple attempts to delete the gene encoding this transpeptidase were fruitless, suggesting it is essential. However, further analysis of srtA was hampered by the lack genetic tools available for DNA manipulation in this organism. To overcome this, our lab has developed facile techniques for generating A. oris conditional mutants and studying the basis for gene essentiality. Using a combination of transcriptional and posttranscriptional mechanisms to control gene expression, we showed that depletion of A. oris srtA results in severe morphological abnormalities and aberrant division septa. To elucidate the pathway for essentiality, our lab then developed a Tn5 transposon system to identify suppressors of the conditional srtA deletion mutant. We discovered that the depletion of this gene resulted in a toxic accumulation of GspA, a glycosylated SrtA substrate, in the membrane. In this chapter, we describe the methods for gene deletions and Tn5-based mutagenesis in A. oris. Protocols for examining the cell morphologies of conditional deletion mutants are also provided.
1.1 Allelic Exchange in A. oris
To avoid potential polar effects by insertion mutations of genes, two methods were developed that generate markerless, in-frame deletion mutants in A. oris. In these methods, the suicide plasmids pCWU2 [10] and pCWU3 [11] were used that express GalK, which phosphorylates d-galactose to generate galactose-1-phosphate, and mCherry as counterselection markers, respectively. To create the deletion constructs, approximately 1 kB regions upstream and downstream of a target gene are PCR amplified, fused together, and then cloned into pCWU2 or pCWU3. The resulting plasmids are electroporated into A. oris and integrated into the chromosome via a single crossover event. To excise target genes from A. oris, a second homologous recombination event is induced by growing the co-integrant strains in the absence of selective antibiotics. When using pCWU2, excision of the plasmid is selected by growing bacteria with 2-deoxy-d-galactose (2-DG), which is converted into a toxic intermediate by GalK. For pCWU3, plasmid excision is selected by loss of cell fluorescence [11].
1.2 Generation of Conditional Deletion Mutants in A. oris
Approximately 20 % of bacterial genes are required for growth and survival [12]. Deletion of these genes in many organisms is sometimes a daunting task. In A. oris, removal of an essential gene from the bacterial chromosome can be achieved by providing a copy of this gene ectopically. For example, to create a conditional srtA deletion mutant, a tetracycline-inducible expression system (P tet ) was utilized [13]. However, the P xyl/tetO promoter of this system is leaky [14]. To provide a posttranscriptional level of gene regulation, we introduced a theophylline-responsive synthetic riboswitch element E* into the P tet system [15]. E* is composed of a small sequence whose secondary RNA structure inhibits protein translation by blocking ribosome binding. Inhibition of translation by E*, however, is relieved upon the addition of theophylline. This additional level of control was used for generation of the conditional srtA deletion mutant [6].
1.3 Random gene Disruptions by Tn5-Based Transposition
To screen for srtA depletion suppressors, we developed a highly efficient Tn5 system for A. oris [6, 16]. This system is based on EZ-Tn5, an in vitro transposon widely used for bacterial mutagenesis [16]. A kanamycin (Kan) resistance gene cassette derived from the Actinomyces/E. coli shuttle plasmid pJRD215 was cloned into pMOD-2<MCS>. The cassette is flanked by 19 bp mosaic ends (ME), which are recognized by EZ-Tn5 transposase for random insertion into the bacterial genome. The newly generated DNA fragment was then PCR amplified, combined with EZ-Tn5 transposase, and electroporated into A. oris.
2 Materials
2.1 Plasmids (Fig. 1)

Vectors used for genetic manipulation of A. oris—(a–c) Derived from pHTT177, pCWU2 and pCWU3 are two suicide plasmids in A. oris. (d–e) pJRD-Sm is a derivative of the Actinomyces/E. coli shuttle vector pJRD215. MCS indicates multiple cloning sites; Kan for kanamycin and Sm for streptomycin. (f) An unmarked, in-frame deletion mutant of A. oris was obtained using pCWU3 by screening for the loss of fluorescence; adapted from Wu and Ton-That [11]
-
1.
pHTT177, a derivative of pUC19 with a Kan resistance gene derived from pJRD215 in place of the original ampicillin (Amp) resistance gene [4].
-
2.
pCWU2, a derivative of pHTT177 containing galK under the control of the rpsJ promoter [10].
-
3.
pCWU3, a derivative of pHTT177 containing rfp (mCherry) under the control of the rpsJ promoter [11].
-
4.
pJRD215, an Actinomyces/E. coli shuttle vector containing Kan and streptomycin (Sm) resistance gene cassettes [17].
-
5.
pJRD-Sm, a derivative of pJRD215 containing only the Sm resistance gene cassette.
2.2 Preparation of A. oris Competent Cells
-
1.
A. oris MG-1 ΔgalK.
-
2.
Heart infusion agar (HIA) plates.
-
3.
Heart infusion broth (HIB).
-
4.
15 % glycine in HIB.
-
5.
Sterile water.
-
6.
Sterile 10 % glycerol.
-
7.
37 °C water bath shaker.
-
8.
37 °C, 5 % CO2 incubator.
-
9.
Disposable culture glass tubes.
-
10.
125 mL Erlenmeyer flask.
-
11.
50 mL centrifuge tubes with printed graduations.
-
12.
Spectrophotometer and plastic cuvettes for measuring cell density at 600 nm (A600).
-
13.
Sterile 1.5 mL centrifuge tubes.
-
14.
Dry ice–ethanol bath.
2.3 Electroporation
-
1.
A. oris competent cells.
-
2.
Plasmid or transposon DNA.
-
3.
HIB.
-
4.
HIA plates with 50 μg/mL Kan.
-
5.
0.2 cm electroporation cuvettes.
-
6.
Electroporator.
-
7.
Sterile 1.5 mL centrifuge tubes.
-
8.
Sterile spreader.
-
9.
37 °C incubator with 5 % CO2 (see Note 1 ).
2.4 Allelic Replacement
-
1.
Phusion® High-fidelity DNA polymerase.
-
2.
DNA oligonucleotide Primers.
-
3.
Agarose gel matrix.
-
4.
DNA extraction kit (for gel purification) (see Note 2 ).
-
5.
T4 DNA ligase and buffer.
-
6.
Restriction enzymes.
-
7.
E. coli DH5α competent cells.
-
8.
Luria broth (LB) and agar.
-
9.
5 M 2-deoxy-d-galactose (2-DG) dissolved in HIB.
-
10.
42 °C water bath.
-
11.
37 °C incubators with and without 5 % CO2.
-
12.
Olympus X171 inverted microscope with TRITC filter FluorChem Q imaging system (Alpha Innotech).
2.5 Conditional srtA Deletion in A. oris
-
1.
Anhydrotetracycline hydrochloride (AHT) suspended in methanol.
-
2.
Theophylline dissolved in HIB.
-
3.
HIA plates with 50 μg/mL Kan and 50 μg/mL Sm.
-
4.
HIA plates with 100 ng/mL AHT and 2 mM theophylline.
-
5.
Additional reagents and equipment listed above.
2.6 Tn5 Transposition
-
1.
pMOD-2<MCS>® (Epicentre).
-
2.
T4 polynucleotide kinase.
-
3.
100 mM Adenosine 5′-triphosphate (ATP).
-
4.
Phusion® high fidelity DNA polymerase.
-
5.
EZ-TN5™ Transposase from Epicentre.
-
6.
Topo blunt-ending cloning kit.
-
7.
0.2 cm electroporation cuvettes.
-
8.
Electroporator.
2.7 Thin Section and Electron Microscopy
-
1.
Sterile 1.5 mL centrifuge tubes.
-
2.
Sterilized water.
-
3.
0.1 mM NaCl.
-
4.
Phosphate buffer saline (PBS).
-
5.
10 % formalin.
-
6.
Glutaraldehyde.
-
7.
Sodium borohydride.
-
8.
Ethanol.
-
9.
Millonig’s buffer (EMS; Hatfield, PA).
-
10.
LR (London Resin) white resin.
-
11.
Oven.
-
12.
Rotator.
-
13.
BEEM® Capsules.
-
14.
Diatome diamond knife.
-
15.
Leica Ultracut microtome.
-
16.
Formvar-carbon-coated 200-mesh nickel grids.
-
17.
1 % uranyl acetate (UA) for negative staining.
-
18.
Filter paper.
-
19.
Dumont tweezers.
-
20.
Transmission Electron Microscope (TEM).
3 Methods
3.1 Construction of Deletion Plasmids
-
1.
Design two sets of primers to amplify ~1 kb regions upstream and downstream of your target gene. As a reference, refer to Table 1 for the design of srtA deletion primers (srtA is annotated as ana_2245 at www.oralgen.org) (see Note 3 ). For a DNA template, isolate A. oris genomic DNA (see Note 4 ).
Table 1 Primers used for constructing srtA conditional mutant -
2.
Purify the resulting PCR products using a DNA gel purification kit. Treat the PCR products with the appropriate restriction enzymes, and ligate the fragments into pCWU2 or pCWU3, precut with the appropriate restriction enzymes (see Table 1).
-
3.
Transform E. coli DH5α with the resulting plasmid. Extract the plasmid from the positive clones and verify the insert.
3.2 Preparation of A. oris Competent Cells
-
1.
Streak A. oris MG-1 or ΔgalK from a frozen stock on a HIA plate, and incubate 2 days at 37 °C with 5 % CO2 incubator to obtain single colonies (see Note 3 ).
-
2.
Inoculate a colony of A. oris into 6 mL of HIB, and incubate overnight at 37 °C with minimal shaking. The next day, dilute 5 mL of the overnight culture into 65 mL of fresh HIB. Grow cells at 37 °C until the OD600 reaches approximately 0.6. Add 25 mL of prewarmed 15 % glycine in HIB to the culture, and incubate at 37 °C for 1 h.
-
3.
Transfer the culture to centrifuge tubes and chill on ice for at least 10 min. Harvest the cell pellet by centrifugation at 4 °C, and discard the supernatant. Wash the cell pellet twice with 30 mL of prechilled 10 % glycerol. Harvest the cell pellet by centrifugation at 4 °C, resuspend the bacteria in 1 mL of 10 % cold glycerol, and then aliquot into prechilled 1.5 mL centrifuge tubes (~200 μL each).
-
4.
Snap-freeze the samples using a dry ice–ethanol bath, and store at −80 °C.
3.3 Electroporation of A. oris with pCWU2 or pCWU3
-
1.
Add at least 1 μg of plasmid, which harbors a deletion construct (ΔgeneX), to an aliquot of A. oris competent cells thawed on ice, and then leave on ice for at least 10 min. Transfer the contents of the tube to a prechilled 0.2 mm cuvette.
-
2.
Electroporate A. oris using the following conditions: Voltage = 2.5 kV, Resistance = 400 Ω, Capacity = 25 μF.
-
3.
Following electroporation, immediately add 1 mL of prewarmed HIB to the cuvette, transfer the contents to a sterile 1.5 mL centrifuge tubes, and recover the cells for 2-3 h at 37 °C with minimal shaking.
-
4.
Harvest the cell pellet by centrifugation, and remove 1 mL of the supernatant. Resuspend the cell pellet in the remaining HIB, and spread the cells onto HIA plates containing 50 μg/mL of Kan.
-
5.
Incubate the plates at 37 °C with 5 % CO2 for approximately 3 days. Colonies that appear after that time should contain a copy of pCWU2 or pCWU3 that is integrated into the chromosome.
3.4 Allelic Exchange Using galK (pCWU2) as a Counterselection Marker
-
1.
To promote excision of pCWU2-ΔgeneX by a double crossover event, dilute an overnight culture of the co-integrant 1:50 into HIB without Kan, and incubate at 37 °C with minimal shaking for 24 h.
-
2.
The following day, dilute the culture 1:100, spread onto HIA containing 0.25 % 2-DG, and incubate the plates at 37 °C with 5 % CO2 for approximately 3 days.
-
3.
Patch at least 20 colonies onto HIA plates with and without Kan, and incubate overnight at 37 °C in a 5 % CO2 incubator. Select at least ten colonies that are sensitive to Kan (i.e., have lost pCWU2), and screen for the loss of the target gene by PCR.
3.5 Allelic Exchange Using mCherry (pCWU3) as a Counterselection Marker
-
1.
To promote excision of pCWU3-ΔgeneX by a double crossover event, dilute an overnight culture of the co-integrant into HIB without Kan, and incubate at 37 °C overnight with minimal shaking. Repeat seven times (see Note 5 ).
-
2.
Dilute the final culture 1/10,000, spread onto HIA plates, and incubate at 37 °C with 5 % CO2 for approximately 3 days.
-
3.
Colonies that have lost pCWU3 should no longer be fluorescent. Screen these cells using a FluorChem Q imaging system with a Cy3 filter.
-
4.
Patch the nonfluorescent colonies onto HIA with and without Kan. Bacteria that have lost pCWU3 should be Kan-sensitive. Screen for the loss of the target gene by PCR.
3.6 Generation of the A. oris Conditional srtA Deletion Mutant
Described here is a general protocol for generating srtA conditional mutants in A. oris, starting with a merodiploid strain, in which the deletion vector pCWU2-ΔsrtA has been integrated into the ΔgalK chromosome. Also used is pTetR-Ω-SrtA, a derivative of pJRD-Sm, in which srtA expression is tightly regulated under the control of a tetracycline-inducible promoter and a theophylline-responsive riboswitch (see Note 6 ) [6]. In principle, this protocol is applicable for any essential gene.
-
1.
Incubate approximately 0.1 μg of pTetR-Ω-SrtA with the competent merodiploid cells on ice for at least 10 min. Transfer the sample into a prechilled cuvette, and electroporate. Following electroporation, immediately add 1 mL of pre-warmed HIB without antibiotics, and transfer the cuvette contents into a 1.5 mL centrifuge tube. Incubate the cells for 2–3 h at 37 °C without shaking. Next, take a 100 μL aliquot of the culture, and spread it on HIA containing 50 μg/mL of Kan and 50 μg/mL Sm. Incubate the plates at 37 °C with 5 % CO2. Colonies would be visible after 2 days of growth.
-
2.
Pick 2 isolated colonies and patch them on agar plates containing Kan and Sm (50 μg/mL). After 3 days of growth, inoculate cells from the two patches in 6 mL of HIB supplemented with 50 μg/mL Kan and Sm for overnight growth at 37 °C.
-
3.
Harvest cells by centrifugation from 100 μL aliquots of the overnight cultures, and collect the cell pellet by centrifugation. Wash the cells in HIB and then resuspend them into 6 mL of HIB containing 50 μg/mL Sm, 100 ng/mL AHT (see Note 7 ), and 2 mM theophylline (see Note 8 ).
-
4.
To select for clones with plasmid excision, plate 1/100 dilutions of the overnight cultures above onto HIA plates containing 0.25 % 2-DG, 100 ng/mL AHT, and 2 mM theophylline. Incubate the plates at 37 °C with 5 % CO2 for approximately 3 days.
-
5.
Double-patch at least 20 colonies onto HIA plates supplemented with 50 μg/mL Sm, 100 ng/mL AHT, and 2 mM theophylline, as well as HIA containing 50 μg/mL Kan, 50 μg/mL Sm, 100 ng/mL AHT, and 2 mM theophylline. Colonies with pCWU2 excision should be sensitive to Kan.
-
6.
Screen at least ten colonies for the loss of the chromosomal srtA gene by PCR.
-
7.
If deletion mutants are not obtained, repeat steps 4–6.
3.7 Construction of the Tn5 Transposon Plasmid pMOD-2/ Kan215
In our previous study, Tn5 transposon mutagenesis was used to find suppressors for srtA lethality [6]. This transposon approach can also be employed in high throughput screens for virulence factors in A. oris.
-
1.
PCR-amplify the Kan resistance gene cassette from the shuttle plasmid pJRD215 (Table 2), and clone it into the MCS of pMOD-2<MCS>, which carries 19 bp mosaic ends (ME).
Table 2 The primer used for Tn5 transposition assay -
2.
To prepare the transposon, phosphorylate ME plus primers (ME plus 9-3 and ME plus 9-5′) with T4 DNA kinase (see Table 2).
-
3.
PCR amplify the ME and Kan resistance gene fragment (kanR) from pMOD-2/Kan215 using the phosphorylated ME plus primers and Phusion® DNA polymerase. The recommended PCR program is provided: 98 °C for 5 min, 30 cycles of 98 °C for 10 s and 55 °C for 20 s and 72 °C for 1 min, 72 °C for 10 min.
3.8 Production of the Tn5 Transposome
-
1.
Prepare the transposome: 2 μL ME-kanR-ME PCR product, 4 μL EZ-Tn5 transposase (Epicentre), 2 μL 100 % glycerol. Incubate the reaction for 2 h at room temperature, and then store at −20 °C.
-
2.
Add approximately 1.5 μL of transposome to the A. oris competent cells. Incubate on ice for at least 10 min. Transfer the mixture to a cuvette for electroporation. Following electroporation, immediately add 1 mL of prewarmed HIB, and recover the cells in a 1.5 mL centrifuge tube at 37 °C for 3 h without shaking. Spread 40 μL aliquots of transposon-treated cultures onto HIA plates (approximately 30) with 50 μg/mL Kan and 50 μg/mL Sm. As a control, spread another aliquot onto HIA containing both antibiotics and 100 ng/mL AHT and 2 mM theophylline (see Note 9 ).
-
3.
Inoculate plates into 37 °C with 5 % CO2 for 3 days.
3.9 Characterization of Tn5 Insertion by TAIL-PCR
Thermal asymmetric interlaced PCR (TAIL-PCR) is a useful tool to map out Tn5 insertion sites [18]. TAIL-PCR is performed by alternate rounds of low stringency and high stringency PCR cycles using transposon-specific and degenerate primers. Below is a simplified TAIL-PCR protocol to determine Tn5 transposon insertion sites in A. oris.
-
1.
First round of PCR (PCR-1): Tn5-1 and AD1 primers (Table 2), A. oris Tn5 suppressor chromosomal DNA as a template, PCR parameters (98 °C for 5 min, 30 cycles of 98 °C for 15 s and 45 °C for 20 s and 72 °C for 1 min, 72 °C for 10 min).
-
2.
Second round of PCR (PCR-2) (see Note 10 ): Tn5-2 and AD2 primers (see Table 2), 1 μL of unpurified product from PCR-1 as a template, PCR parameters (98 °C for 5 min, 30 cycles of 98 °C for 15 s and 60 °C for 20 s and 72 °C for 1 min, 72 °C for 10 min).
-
3.
Third round of PCR (PCR-3): Unpurified products from PCR-2 are used for PCR-3 reactions with conditions similar to PCR-2.
-
4.
Clone the products into a TOPO blunt-end cloning vector, and sequence the vector containing PCR-3 product using primers M13F/M13R (see Table 2) to identify the site of Tn5 insertion (see Note 11 ).
3.10 Preparation of A. oris Thin Sections
For high-resolution studies of bacterial morphology, thin-section electron microscopy is an excellent choice of methodology. Described here is a protocol for preparing A. oris thin sections.
-
1.
Scrap A. oris cells grown from HIA plates and suspend in 1 mL of PBS. Harvest cells by centrifugation and resuspend them in a fixative solution (3 % formalin, 0.15 % glutaraldehyde in Millonig’s buffer, pH 7.4). Incubate for 4 days at 4 °C.
-
2.
After harvesting cells centrifugation, wash them in fresh 0.1 % borohydride in Millonig’s buffer for 10 min, followed by washing three times in Millonig’s buffer alone for 5 min. Harvest the cells by centrifugation and wash them in water for 5 min.
-
3.
Dehydrate the samples with graded concentrations of cold 50 % methanol for 5 min three times, followed by dehydration with cold 70 % methanol three times.
-
4.
Infiltrate with 50 % LR White resin (EMS; Hatfield, PA)–50 % methanol mixtures at −20 °C for 2 h. Repeat this step once, followed by infiltration with 75 % LR white resin: 25 % methanol mixtures at −20 °C for 4 h. Finally, infiltrate with 100 % LR white resin on rotation at room temperature overnight.
-
5.
Embed the pellets in BEEM capsules and polymerize in a 50 °C oven for 2 days.
-
6.
Cut approximately 50 nm thin sections with a diamond knife using an ultramicrotome, and place onto Formvar-carbon-coated 200-mesh nickel grids.
3.11 Transmission Electron Microscopy (TEM)
-
1.
Drop 10 μL of 0.25 % uranyl acetate on top of a grid containing the A. oris thin sections, and incubate at room temperature for 1 min (see Note 12 ).
-
2.
Wick excess uranyl acetate using filter paper.
-
3.
View the samples by TEM (Fig. 2).
Fig. 2 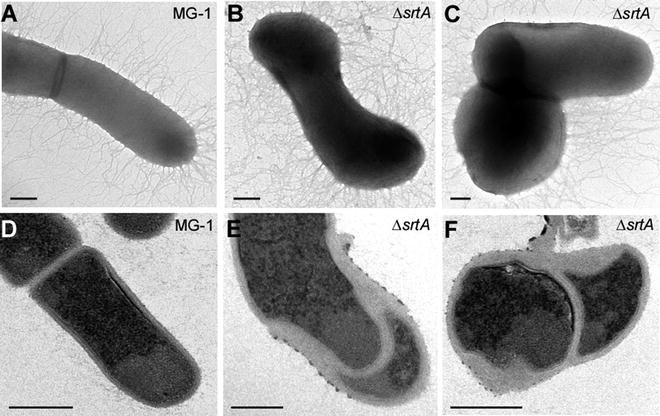
Electron microscopic analysis of A. oris—Cells from the A. oris parental strain MG-1 (a and d) and srtA-depleted cells (b, c, e, and f) were examined by electron microscopy. Scale bars indicate 0.2 μm

4 Notes
-
1.
If a CO2 incubator is not available, wrap the HIA plates in Parafilm to limit their exposure to oxygen.
-
2.
Qiagen kits are available for purchase.
-
3.
Note that the upstream and downstream PCR products are fused by restrictions sites. This could also be achieved by overlapping PCR.
-
4.
Genomic DNA can be isolated using phenol–chloroform extraction or a Promega Wizard® Genomic DNA purification kit.
-
5.
The efficiency of galk-based counterselection is higher than the mCherry-based method. However, the mCherry counterselection does not require the A. oris ΔgalK background.
-
6.
The riboswitch element E* sequence (The RBS is highlighted in grey):
GGUACC GGUGAUACCAGCAUCGUCUUGAUGCCCUUGGCAGCACCCUGCUAAGGAGGCAACAAGAUG
-
7.
These are necessary to induce the plasmid-borne srtA. The absence of Kan will select for excision of the chromosomal copy of the gene of interest.
-
8.
Anhydrotetracycline (AHT) is a non-bacteriostatic derivative of tetracycline.
-
9.
Only srtA depletion suppressor mutants will grow in the absence of the inducers AHT and theophylline.
-
10.
The annealing temperature for the second round of PCR can be adjusted from 55 to 65 °C only 1–2 major PCR fragments are obtained.
-
11.
Sequences positive for Tn5 insertion should contain ME sequences.
-
12.
Residual PBS will react with uranyl acetate resulting in poor visibility of the samples by TEM.
References
Jakubovics NS, Kolenbrander PE (2010) The road to ruin: the formation of disease-associated oral biofilms. Oral Dis 16(8):729–739
Chen L, Ma L, Park NH, Shi W (2001) Cariogenic actinomyces identified with a beta-glucosidase-dependent green color reaction to Gardenia jasminoides extract. J Clin Microbiol 39(8):3009–3012
Wu C, Mishra A, Yang J, Cisar JO, Das A, Ton-That H (2011) Dual function of a tip fimbrillin of Actinomyces in fimbrial assembly and receptor binding. J Bacteriol 193(13):3197–3206
Mishra A, Das A, Cisar JO, Ton-That H (2007) Sortase-catalyzed assembly of distinct heteromeric fimbriae in Actinomyces naeslundii. J Bacteriol 189(8):3156–3165
Reardon-Robinson ME, Wu C, Mishra A, Chang C, Bier N, Das A, Ton-That H (2014) Pilus hijacking by a bacterial coaggregation factor critical for oral biofilm development. Proc Natl Acad Sci U S A 111(10):3835–3840
Wu C, Huang IH, Chang C, Reardon-Robinson ME, Das A, Ton-That H (2014) Lethality of sortase depletion in Actinomyces oris caused by excessive membrane accumulation of a surface glycoprotein. Mol Microbiol 94(6):1227–1241
Mazmanian SK, Ton-That H, Schneewind O (2001) Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol Microbiol 40(5):1049–1057
Novick RP (2000) Sortase: the surface protein anchoring transpeptidase and the LPXTG motif. Trends Microbiol 8(4):148–151
Bradshaw WJ, Davies AH, Chambers CJ, Roberts AK, Shone CC, Acharya KR (2015) Molecular features of the sortase enzyme family. FEBS J 282(11):2097–2114
Mishra A, Wu C, Yang J, Cisar JO, Das A, Ton-That H (2010) The Actinomyces oris type 2 fimbrial shaft FimA mediates co-aggregation with oral streptococci, adherence to red blood cells and biofilm development. Mol Microbiol 77(4):841–854
Wu C, Ton-That H (2010) Allelic exchange in Actinomyces oris with mCherry fluorescence counterselection. Appl Environ Microbiol 76(17):5987–5989
Christen B, Abeliuk E, Collier JM, Kalogeraki VS, Passarelli B, Coller JA, Fero MJ, McAdams HH, Shapiro L (2011) The essential genome of a bacterium. Mol Syst Biol 7:528
Carroll P, Muttucumaru DG, Parish T (2005) Use of a tetracycline-inducible system for conditional expression in Mycobacterium tuberculosis and Mycobacterium smegmatis. Appl Environ Microbiol 71(6):3077–3084
Corrigan RM, Foster TJ (2009) An improved tetracycline-inducible expression vector for Staphylococcus aureus. Plasmid 61(2):126–129
Topp S, Reynoso CM, Seeliger JC, Goldlust IS, Desai SK, Murat D, Shen A, Puri AW, Komeili A, Bertozzi CR, Scott JR, Gallivan JP (2010) Synthetic riboswitches that induce gene expression in diverse bacterial species. Appl Environ Microbiol 76(23):7881–7884
Goryshin IY, Reznikoff WS (1998) Tn5 in vitro transposition. J Biol Chem 273(13): 7367–7374
Yeung MK, Kozelsky CS (1994) Transformation of Actinomyces spp. by a gram-negative broad-host-range plasmid. J Bacteriol 176(13): 4173–4176
Liu YG, Whittier RF (1995) Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 25(3):674–681
Acknowledgments
This work was supported by the National Institute of Dental and Craniofacial Research of the NIH under award numbers F31DE024004 (to M.E.R.-R.) and DE017382 and DE025015 (to H.T.-T.).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Wu, C., Reardon-Robinson, M.E., Ton-That, H. (2016). Genetics and Cell Morphology Analyses of the Actinomyces oris srtA Mutant. In: Hong, HJ. (eds) Bacterial Cell Wall Homeostasis. Methods in Molecular Biology, vol 1440. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3676-2_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3676-2_9
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3674-8
Online ISBN: 978-1-4939-3676-2
eBook Packages: Springer Protocols