
Abstract
Cancer cells are characterized by rapid proliferation and require adaptive metabolic responses to allow continued biosynthesis and cell growth in the setting of decreased oxygen (O2) and nutrient availability. The hypoxia-inducible factors (HIFs) are a common link between adaptation to low O2, changes in cancer metabolism, and malignant progression. The HIF-α subunits differentially regulate metabolic enzymes and other key factors involved in glycolysis, changes in redox status, and oxidative phosphorylation. Importantly, metabolic changes can, in turn, regulate HIF activity. Finally, changes in metabolism under hypoxia lead to important crosstalk between cancer cells and the stromal compartment of the microenvironment.
Similar content being viewed by others


Hypoxia-inducible factors and their regulation
A majority of healthy tissues experience 2–9 % O2, while hypoxia is defined as less than 2 % O2 [1]. Low O2 tensions are often exhibited by regions of intense inflammation such as within arthritic joints, regions of the bone marrow, and in highly proliferative cancer cells [2]. Hypoxia may occur secondary to necrosis or aberrant neovascularization resulting in poor perfusion. Additionally, cancer cells may also proliferate rapidly enough to outstrip their blood supply [3]. Cells adapt to changes in O2 availability by altering gene expression of crucial metabolic enzymes in order to counter changes in nutrient availability and redox status. This response is mediated, in part, by O2-labile transcription factors hypoxia-inducible factors HIF-1α and HIF-2α, key regulators of cellular adaptation to hypoxic stress [3–5].
Comprised of an O2-sensitive α subunit and constitutively expressed β subunit, HIFs are primarily regulated through post-translational modification and stabilization and are part of the basic helix-loop-helix-PAS (bHLH/PAS) family of transcription factors [6]. Under normal O2 tensions, prolyl hydroxylase domain enzymes (PHDs) hydroxylate two conserved proline residues (405 and 531 in HIF-1α) within the O2-dependent degradation (ODD) domain of the HIF-α subunit. After hydroxylation, the von Hippel-Lindau (VHL) tumor suppressor E3 ligase complex polyubiquitinates HIF-α and targets it for eventual degradation by the 26S proteasome [7–9]. Under low O2, HIFs are no longer modified by PHDs, but instead dimerize with ARNT/HIF-1β through HLH and PAS domain interactions, translocate to the nucleus, and recruit coactivators such as CBP/p300. HIF heterodimers bind and recognize hypoxia-response elements (HREs), with the consensus sequence G/ACGTG, within the promoter regions of target genes and drive adaptive gene transcription [10–13] (Fig. 1). While the HIF-1α subunit is expressed ubiquitously, HIF-2α is selectively expressed in a much more tissue-restricted manner but can be found at high levels in vascular endothelial cells and myeloid-derived cells [14].

Regulation of hypoxia-inducible factors. Under normoxia, the alpha subunit of the hypoxia-inducible factors (HIF) gets hydroxylated in a prolyl hydroxylase (PHD)-dependent manner. The von Hippel-Lindau E3 ubiquitin ligase (VHL) then ubiquitilates HIF-α, thus tagging it for proteasome-mediated degradation. Under hypoxia, HIF-α subunits are stabilized, bind their obligate partner ARNT, translocate into the nucleus where the HIF-α/ARNT dimer binds to hypoxic response elements (HREs) on target gene promoters and activates transcription of several genes
Cancer cells maintain rapid proliferation, even in the absence of adequate O2 and nutrient availability. Adaptive metabolic changes allow continued biosynthesis and cell growth in the setting of decreased access to O2. As cancer cells divide and outstrip O2 and nutrient supplies, HIF activation allows modulation of cell metabolism and gene expression to adapt to the ever-changing landscape of the tumor microenvironment. HIF expression is an important prognostic factor, as levels associate closely with poor patient outcomes in a variety of cancers [13]. While both factors share certain downstream transcriptional targets, they exhibit different roles in various aspects of cell metabolism and proliferation [15]. In this review, we will characterize some of the distinct pathways that allow for the alteration of cancer metabolism in response to hypoxia.
Effects of HIF activity on tumor cell metabolism
In 1924, Otto Warburg made the observation that cancer cells uptake high levels of glucose to produce lactic acid even under high O2 concentrations (aerobic glycolysis). This finding, termed the “Warburg effect”, is a critical feature of solid tumors and the basis of diagnostic cancer imaging techniques, such as fluorodeoxyglucose positron emission tomography (FDG-PET) [16]. Largely until now, the molecular basis of the Warburg effect and rationale behind cancer cells undergoing aerobic glycolysis have not been completely elucidated [17].
Glycolysis allows for continued ATP production without the need for O2-dependent oxidative phosphorylation, and is thus an important pathway under hypoxic conditions. Metabolic intermediates from the glycolytic pathway can also be funneled into macromolecular biosynthesis for continued growth. HIF-1α activates transcription of genes encoding glucose transporters and other glycolytic enzymes such as LDHA, PGK-1, and HK1 and thus plays an important role in the glycolytic switch [17]. The HIFs prevent entry into the TCA cycle in part by preventing acetyl coenzyme A (acetyl-CoA) production from pyruvate, and thus redirect glucose away from the oxidative phosphorylation pathway. Pyruvate dehydrogenase (PDH), the enzyme responsible for conversion of pyruvate to acetyl-CoA, is indirectly regulated by HIF activity through the modifying enzyme pyruvate dehydrogenase kinase (PDK). PDK1 is upregulated by HIF-1α expression and in turn deactivates PDH. Hypoxia and HIF increase glucose/carbon flux through the glycolytic pathway while minimizing input into the TCA cycle and oxidative phosphorylation [17].
IDH activity and the TCA cycle under hypoxia
Hypoxia and HIFs have been implicated in promoting reverse flux through the TCA cycle and lipogenesis through activity of isocitrate dehydrogenase-1 (IDH1), an enzyme that promotes reductive carboxylation of glutamine to α-ketoglutarate and citrate [18, 19]. Renal cell lines lacking VHL exhibit constitutive reductive carboxylation of glutamine to α-ketoglutarate for de novo lipid biosynthesis at normal O2 tensions, implicating the O2-labile HIF subunits in regulation of this oxygen-sensitive pathway [18, 20]. An obvious role exists for HIF-mediated control of mammalian cell metabolism and acetyl-CoA production for lipogenesis under hypoxia. Cells may adjust to low O2 tensions through changes in metabolism and methods of ATP production, but are also known to adaptively alter strategies for macromolecular biosynthesis, using metabolic intermediates to fuel nucleotide and lipid synthesis. Hypoxic cells have unique metabolic needs and respond through HIF-regulated changes in metabolism and protein synthesis.
Oxygen consumption, HIF-1α and inhibition of oxidative phosphorylation
Decreasing the amount of O2 consumed under hypoxia is one of the central metabolic adaptations in tumor microenvironments. Most O2 consumption in cells is dedicated to oxidative phosphorylation (OXPHOS) in the mitochondria [21]. Under low O2 tensions, HIF-1α mediates the switch from OXPHOS to anaerobic glycolysis by the upregulation of a series of genes that lead to active uptake of glucose and inhibition of the TCA cycle. As previously stated, hypoxia-induced PDH inactivation ultimately leads to a shift away from the TCA cycle [22]. To control for efficient O2 use and prevent harmful accumulation of reactive oxygen species (ROS), HIF-1α transcriptionally regulates genes that control the content of the electron transport chain (ETC) complexes in the mitochondria. For example, HIFs have been shown to regulate cytochrome c oxidase subunits by activating the transcription of cytochrome oxidase 4-2 (COX4-2) and LON, the mitochondrial protease responsible for degrading the COX4-1 isoform [23]. The switch of COX4-1 for COX4-2 leads to more efficient aerobic respiration during hypoxia, and protects the cells from elevated ROS production.
Yet, there are additional unknown mechanisms through which hypoxia and hypoxic responses can lead to the attenuation of mitochondrial function for OXPHOS. A recent study identified a novel HIF-1α target gene which inhibits mitochondrial complex I activity: NADH dehydrogenase [ubiquinone] 1 alpha subcomplex 4-like 2 (NDUFA4L2) [24]. NDUFA4L2 was shown to inhibit complex I activity in both normal and tumor cells, suggesting that NDUFA4L2-mediated complex I inhibition is one of the many mechanisms through which cells adapt to low O2 tension. Importantly, silencing of NDUFA4L2 also leads to increased ROS production and ultimately impaired cellular proliferation, making NDUFA4L2 a crucial mediator of survival adaptation under hypoxic stress [24]. Taken together, these findings underscore the importance of HIFs controlling O2 consumption in hypoxic cells through many independent mechanisms.
HIF-1α–PKM2 connections
The M2 isoform of pyruvate kinase (PKM2) is a glycolytic enzyme that is expressed by highly proliferating cells and converts phosphoenolpyruvate (PEP) into pyruvate. Several recent findings place this enzyme at the crossroads between glucose utilization for ATP production or cellular proliferation (reviewed in [25]). Therefore, a rapid evolution in understanding the biology of PKM2 has occurred, beginning with the finding that this enzyme is negatively regulated by tyrosine phosphorylation, to the elucidation of a pro-proliferative role for the inactive form of PKM2. Moreover, PKM2 function in the nucleus is currently being established.
A recent study has established a novel connection between PKM2 and HIF-1α that is independent of its enzymatic properties. Luo and colleagues [26] have demonstrated that HIF-1α and PKM2 physically interact in the nucleus, positively modulating HIF-1α transcriptional activity. Importantly, PKM2 has been previously established to be a transcriptional target of HIF-1α [27]. Moreover, the authors show that the PKM2–HIF-1α interaction is strengthened through a prolyl hydroxylase 3 (PHD3)-mediated hydroxylation of PKM2 [26]. Taken together, these findings suggest that PKM2 plays an important role in malignant metabolic reprogramming under low O2 conditions, by enhancing the pro-glycolytic signals controlled by HIF-1α [28]. While it still remains to be tested how widely this interaction occurs in cancer, this new finding elucidates a novel connection between glucose metabolism, hypoxia, and tumorigenesis.
Hypoxic changes in cell metabolism and HIF-1α/HIF-2α differential regulation
Researchers continually discover additional mechanisms of HIF regulation. For example, the metabolic profile of a cell can regulate HIF activity. The deacetylase sirtuin 1 (Sirt1), a known stress-response factor, has been implicated in HIF regulation. Sirt1 is activated by changes in redox balance that occur as a result of hypoxia-induced metabolic changes (as previously mentioned) and can selectively deacetylate specific lysine residues in HIF-1α and HIF-2α in a hypoxia-dependent manner. HIF-1α and HIF-2α exhibit differential interactions with Sirt1 resulting in opposing downstream effects. Under normal O2 tensions Sirt1 deacetylates HIF-1α and prevents recruitment of the cofactor p300, thereby inhibiting downstream target genes. The opposite is true for the effects of Sirt1 on HIF-2α. Thus, as is often the case with the HIFs, there appears to be opposite effects from acetylation [29]. Sirt1 has been shown to augment HIF-2α regulation of erythropoietin [30]. Because Sirt1 is a stress-induced deacetylase, there is the intriguing possibility of non-hypoxic HIF regulation. Sirt1 responds to changes in the ratio of NAD+ to NADH, which may be altered by influences other than hypoxia, such as nutrient deprivation. If redox change is prevented by inhibiting glycolysis, even under hypoxia, Sirt1 is upregulated and results in HIF-1α inhibition [31]. Thus, even under hypoxia, HIF-1α is not stabilized but is instead inhibited through lysine-acetylation by Sirt1. Together, these observations provide both hypoxia-dependent and -independent roles for Sirt1 regulation of HIF.
HIF and the proto-oncogene c-Myc
Hypoxia and HIFs can also alter tumor metabolism through cooperation with proto-oncogenes that increase cell proliferation and tumor progression. MYC amplification or overexpression occurs in 70 % of all cancers and increases energy and biomolecular synthesis necessary for rapid cellular proliferation [32]. Interestingly, HIF-1α and HIF-2α have differential effects on Myc activity. While HIF-1α stabilization inhibits c-Myc and arrests cellular proliferation, HIF-2α enhances c-Myc activity and induces progression through the cell cycle [15]. Whereas HIF-2α enhances c-Myc-driven cell proliferation, HIF-1α (depending on context) may antagonize c-Myc activity [33, 34]. HIF-1α and HIF-2α have differential roles in c-Myc activity through opposing interactions with c-Myc cofactors, resulting in the stabilization or disruption of c-Myc DNA binding. The predominant HIF-α subunit will therefore affect tumor kinetics and resistance to stress, important factors when considering treatment [34].
HIF–Myc interactions are important, especially in light of Myc’s roles in modulating glucose and glutamine metabolism. Myc overexpression drives proliferation even under energetic stress, such as during glucose deprivation and hypoxia. Myc can cooperate with HIF-1α resulting in increased pyruvate dehydrogenase kinase (PDK1) expression and further inhibition of carbon flux through the TCA cycle and oxidative phosphorylation [17]. However, in the absence of adequate glucose and O2, c-Myc promotes sufficient TCA flux through glutamine metabolism. Recently, Le and colleagues [35] subjected the c-Myc-inducible P493 Burkitt lymphoma cell line to glucose and O2 deprivation, showing a crucial role for c-Myc in shunting glutamine intermediates into the TCA cycle. Thus, c-Myc-controlled glutaminolysis diverts available carbon for further synthesis of macromolecular intermediates. This strengthens the notion that glutamine metabolism is important for cell survival under stress [35]. This finding may also explain why the interaction between HIF-2α and c-Myc is beneficial to malignant proliferation under low O2. Importantly, the role of c-Myc in mediating metabolic changes that confer pro-growth and pro-survival advantages under nutrient deprivation and hypoxia may suggest novel therapeutic approaches for Myc-driven cancers such as Burkitt lymphoma.
Metabolism, oxygen, and tumor–stroma interactions
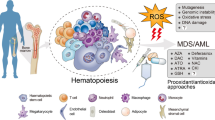
Most of the studies involving cancer metabolism have been tumor cell intrinsic. However, crosstalk between cancer metabolism and the metabolism of non-malignant stroma is also crucial for malignant progression.
Tumor–stroma interactions in FLT inhibitor-resistant AML
Acute myelogenous leukemias (AML) with Fms-like tyrosine kinase-3 (FLT3) mutations frequently evade FLT3 inhibitor therapy, but the mechanism through which this evasion is achieved remains poorly understood [36, 37]. Recent work has shown that AML cells are protected by their interactions with bone marrow stromal cells (BMSCs), thought to be mediated in part by signaling downstream of CXCL12/CXCR4 interactions. FLT3-mutated AML cells express CXCR4, and rely on the availability of CXCL12 from the stroma for proliferation and survival. Since BMSCs reside in a hypoxic microenvironment, HIF-1α is activated and transcriptionally upregulates CXCL12. Overexpression of CXCL12 by stromal cells can be blunted through a p53-mediated HIF-1α degradation [38]. Thus, activation of p53 or inactivation of HIF-1α in stromal cells could lead to a better outcome for patients with FLT3-mutated AML by downregulating the ability of stromal cells to produce CXCL12.
BMSCs provide cysteine availability for CLL cell survival
While it is well established that tumor cells can reprogram their internal metabolism for viability and adaptation under stress conditions, they still require external metabolites for proper survival [39]. A recent study has shown that chronic lymphocytic leukemias (CLL) survive by importing the essential amino acid cysteine from their microenvironment [40].
Due to their uncontrolled proliferation, CLL cells have intrinsically higher levels of ROS compared to normal lymphocytes [41], making them vulnerable to agents that exacerbate ROS production. To maintain redox balance, CLL cells are increasingly dependent on antioxidants such as glutathione (GSH), and freely metabolize glutathione when co-cultured with BMSCs, but lack the ability to obtain this metabolite when cultured alone. Research from Zhang and colleagues [40] has shown that CLL cells are less viable if they are unable to obtain cysteine, a precursor for GSH. Cysteine availability depends on a cell’s ability to uptake cystine (cysteine molecules connected by a disulfide bond) from the microenvironment and converting it into cysteine. Zhang et al. showed that CLL cells do not express high levels of the antiporter responsible for cystine transport into the cell [39, 40]. Non-malignant BMSCs, however, can import cystine, convert it to cysteine, and provide cysteine to CLL cells for GSH synthesis. Blocking cysteine availability, or GSH production, may lead to increased ROS accumulation and eventually, apoptosis of the malignant cells. This new finding underscores the importance of studying hematological malignancies in the context of their tumor microenvironment.
Tumor-associated macrophages experience differential NO metabolism under hypoxia
For example, tumor-associated macrophages (TAMs) are established stromal components of the tumor microenvironment that are often seen associated with regions of intense hypoxia. TAMs adapt to hypoxia through specific changes in gene expression including, but not limited to, upregulation of specific metabolic genes. In addition to changes in gene expression, hypoxia can also result in rapid conversion from aerobic to anaerobic glycolysis [42]. This results from a switch to the more active isoenzyme of phosphofructokinase and an increase in fructose-2,6-bisphosphate concentration and overall glycolytic flux under hypoxic conditions [43]. Additionally HIF-1α induces expression of glucose transporter 1 (GLUT-1) and phosphoglycerate kinase (PGK); macrophages that lack HIF-1α expression have decreased cellular ATP when placed under hypoxia. This change in metabolic status of myeloid-derived cells results in decreased recruitment, as well as decreased migration and invasion [44].
Nitric oxide (NO) metabolism and homeostasis is also of great functional importance for hypoxic macrophages [45]. Similar to previous findings, HIF-1α and HIF-2α have distinct and somewhat opposing roles in NO regulation. Depending on macrophage polarization, NO production is controlled through distinct mechanisms in a HIF-dependent manner. While classically activated, pro-inflammatory macrophages correspond to HIF-1α upregulation as induced by Th1 cytokines, HIF-2α activation is detected in alternatively activated macrophages and induced by Th2 cytokines [45]. When HIF-2α is induced in an alternatively activated macrophage, arginase1 is expressed and suppresses NO production. This contrasts with HIF-1α which, when present in a classically activated macrophage, promotes iNOS expression and increases NO production facilitating removal of pathogens. The HIF-α isoforms are primarily expressed in differently activated macrophages and have opposing roles in NO production which are crucial for the unique functions required by diverse macrophage populations. Thus, NO production and HIF-α isoform expression are closely aligned with macrophage polarization and metabolism [45].
Conclusion
HIFs are O2-sensitive transcription factors that allow transcriptional adaptation to hypoxic environments. It is becoming increasingly apparent that HIFs are common links between O2 availability, malignant progression, and changes in cancer metabolism. HIF-1α and HIF-2α can transcriptionally mediate changes in the levels of metabolic enzymes and other key factors involved in glycolysis, changes in redox status, oxidative phosphorylation, and more. Conversely, changes in key modulators of cancer metabolism can also regulate HIF functions. Importantly, hypoxia and the HIFs also mediate the metabolic crosstalk between malignant cells and the stroma (Fig. 2). Future work is needed to further elucidate the connections between hypoxia and metabolism, especially in the setting of hematologic malignancies. Cancer cell metabolism is an area garnering increasing attention and one that shows great promise in the setting of novel cancer therapy.

Intracellular and tumor microenvironment interactions under hypoxia. The HIF transcription factors regulate hypoxic response in cancer cells by mediating interactions with other transcription factors such as Myc and p53, as well as various metabolic enzymes (PKM2, IDH1/2, etc.). Additionally, cancer cells interact with their microenvironment through cytokine and metabolite signaling (see text). HIF hypoxia-inducible factors, PKM2 M2 pyruvate kinase, OXPHOS oxidative phosphorylation, TCA cycle tricarboxylic acid cycle
References
Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8(12):967–75.
Murdoch C, Muthana M, Lewis CE. Hypoxia regulates macrophage functions in inflammation. J Immunol. 2005;175(10):6257–63.
Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40(2):294–309.
Ema M, et al. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci USA. 1997;94(9):4273–8.
Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11(1):72–82.
Qing G, Simon MC. Hypoxia inducible factor-2alpha: a critical mediator of aggressive tumor phenotypes. Curr Opin Genet Dev. 2009;19(1):60–6.
Cockman ME, et al. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275(33):25733–41.
Jaakkola P, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–72.
Maxwell PH, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–5.
Imtiyaz HZ, et al. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest. 2010;120(8):2699–714.
Talks KL, et al. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157(2):411–21.
White JR, et al. Genetic amplification of the transcriptional response to hypoxia as a novel means of identifying regulators of angiogenesis. Genomics. 2004;83(1):1–8.
Semenza GL. Life with oxygen. Science. 2007;318(5847):62–4.
Patel SA, Simon MC. Biology of hypoxia-inducible factor-2alpha in development and disease. Cell Death Differ. 2008;15(4):628–34.
Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12(2):108–13.
Shanmugam M, McBrayer SK, Rosen ST. Targeting the Warburg effect in hematological malignancies: from PET to therapy. Curr Opin Oncol. 2009;21(6):531–6.
Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20(1):51–6.
Metallo CM, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481(7381):380–4.
Mullen AR, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481(7381):385–8.
Wise DR, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci USA. 2011;108(49):19611–6.
Papandreou I, et al. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3(3):187–97.
Kim JW, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–85.
Fukuda R, et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129(1):111–22.
Tello D, et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting complex I activity. Cell Metab. 2011;14(6):768–79.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324((5930)):1029–33.
Luo W, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145(5):732–44.
Semenza GL, et al. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269(38):23757–63.
Luo W, Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget. 2011;2(7):551–6.
Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22.
Dioum EM, et al. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science. 2009;324(5932):1289–93.
Lim JH, et al. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. 2010;38(6):864–78.
Gordan JD, et al. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11(4):335–47.
Kaelin WG Jr. Kidney cancer: now available in a new flavor. Cancer Cell. 2008;14(6):423–4.
Gordan JD, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14(6):435–46.
Le A, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15(1):110–21.
Zhang W, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. 2008;100(3):184–98.
Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116(24):5089–102.
Kojima K, et al. p53 activation of mesenchymal stromal cells partially abrogates microenvironment-mediated resistance to FLT3 inhibition in AML through HIF-1alpha-mediated down-regulation of CXCL12. Blood. 2011;118(16):4431–9.
Deberardinis RJ. Good neighbours in the tumour stroma reduce oxidative stress. Nat Cell Biol. 2012;14(3):235–6.
Zhang W, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14(3):276–86.
Zhou Y, et al. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003;101(10):4098–104.
Kawaguchi T, Veech RL, Uyeda K. Regulation of energy metabolism in macrophages during hypoxia. Roles of fructose 2,6-bisphosphate and ribose 1,5-bisphosphate. J Biol Chem. 2001;276(30):28554–61.
Rodriguez-Prados JC, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185(1):605–14.
Cramer T, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–57.
Takeda N, et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24(5):491–501.
Author information
Authors and Affiliations
Corresponding author
Additional information
V. Mucaj and J.E.S. Shay contributed equally to this work.
About this article
Cite this article
Mucaj, V., Shay, J.E.S. & Simon, M.C. Effects of hypoxia and HIFs on cancer metabolism. Int J Hematol 95, 464–470 (2012). https://doi.org/10.1007/s12185-012-1070-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-012-1070-5