
Abstract
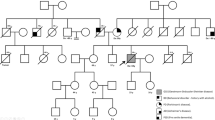
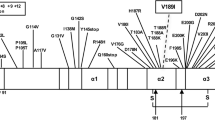
A P102L point mutation in the prion protein gene (PRNP) usually causes Gerstmann–Sträussler–Scheinker disease (GSS), which is a rare hereditary transmissible spongiform encephalopathy (TSE). The clinical features include ataxia in 50s age group with subsequent dementia, spastic paraparesis and extrapyramidal signs. Many families have been reported from the Caucasian population, but only one from the Chinese. We hereby report a large Chinese family with P102L mutation of PRNP whose clinical manifestations at onset were intriguingly heterogeneous, either rapidly progressive dementia with scanty other neurological features or slowly progressive ataxia followed by cognitive impairment. The four-generation pedigree included eight patients with a mean age at onset of 36.9 ± 12.9 (mean ± SD) years. Mean disease duration to death in the four patients was 5.5 ± 1.7 (mean ± SD) years. Molecular analysis revealed a P102L mutation and M129 polymorphism in the PRNP gene in all affected individuals. TSE with P102L mutation of PRNP appears to have a remarkably variable phenotypic expressivity that may change with time and does not appear related to the codon 129 polymorphism.


Similar content being viewed by others

References
Alperovitch A, Zerr I, Pocchiari M, Mitrova E, de Pedro Cuesta J, Hegyi I, Collins S, Kretzschmar H, van Duijn C, Will RG (1999) Codon 129 prion protein genotype and sporadic Creutzfeldt–Jakob disease. Lancet 353:1673–1674
Arata H, Takashima H, Hirano R, Tomimitsu H, Machigashira K, Izumi K, Kikuno M, Ng AR, Umehara F, Arisato T, Ohkubo R, Nakabeppu Y, Nakajo M, Osame M, Arimura K (2006) Early clinical signs and imaging findings in Gerstmann–Straussler–Scheinker syndrome (Pro102Leu). Neurology 66:1672–1678
Barbanti P, Fabbrini G, Salvatore M, Petraroli R, Cardone F, Maras B, Equestre M, Macchi G, Lenzi GL, Pocchiari M (1996) Polymorphism at codon 129 or codon 219 of PRNP and clinical heterogeneity in a previously unreported family with Gerstmann–Straussler–Scheinker disease (PrP-P102L mutation). Neurology 47:734–741
Budka H (2003) Neuropathology of prion diseases. Br Med Bull 66:121–130
Collinge J (2001) Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 24:519–550
Collinge J, Palmer MS (1992) Prion diseases. Curr Opin Genet Dev 2:448–454
Collinge J, Rossor M (1996) A new variant of prion disease. Lancet 347:916–917
Collins S, McLean CA, Masters CL (2001) Gerstmann–Straussler–Scheinker syndrome, fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci 8:387–397
Collins SJ, Lawson VA, Masters CL (2004) Transmissible spongiform encephalopathies. Lancet 363:51–61
Dlouhy SR, Hsiao K, Farlow MR, Foroud T, Conneally PM, Johnson P, Prusiner SB, Hodes ME, Ghetti B (1992) Linkage of the Indiana kindred of Gerstmann–Straussler–Scheinker disease to the prion protein gene. Nat Genet 1:64–67
Doh-ura K, Tateishi J, Sasaki H, Kitamoto T, Sakaki Y (1989) Pro-leu change at position 102 of prion protein is the most common but not the sole mutation related to Gerstmann-Straussler syndrome. Biochem Biophys Res Commun 163:974–979
Folstein MF, Folstein SE, McHugh PR (1975) Mini-mental state. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198
Furukawa H, Kitamoto T, Tanaka Y, Tateishi J (1995) New variant prion protein in a Japanese family with Gerstmann-Straussler syndrome. Brain Res Mol Brain Res 30:385–388
Harder A, Jendroska K, Kreuz F, Wirth T, Schafranka C, Karnatz N, Theallier-Janko A, Dreier J, Lohan K, Emmerich D, Cervos-Navarro J, Windl O, Kretzschmar HA, Nurnberg P, Witkowski R (1999) Novel twelve-generation kindred of fatal familial insomnia from germany representing the entire spectrum of disease expression. Am J Med Genet 87:311–316
Hill AF, Joiner S, Beck JA, Campbell TA, Dickinson A, Poulter M, Wadsworth JD, Collinge J (2006) Distinct glycoform ratios of protease resistant prion protein associated with PRNP point mutations. Brain 129:676–685
Hill AF, Joiner S, Wadsworth JD, Sidle KC, Bell JE, Budka H, Ironside JW, Collinge J (2003) Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain 126:1333–1346
Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, Westaway D, Ott J, Prusiner SB (1989) Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature 338:342–345
Gerstmann ES J, Scheinker J (1936) U˜ ber eine eigenartige hereditär-familiäre Erkrankung des Zentralnervensystems. Zeitschr Gesamte Neurol Psychiatr (Berl) 154:736–762
Jung KY, Seo DW, Na DL, Chung CS, Lee IK, Oh K, Im CH, Jung HK (2007) Source localization of periodic sharp wave complexes using independent component analysis in sporadic Creutzfeldt-Jakob disease. Brain Res 1143:228–237
Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, Collins SJ, Boyd A, Giulivi A, Coulthart M, Delasnerie-Laupretre N, Brandel JP, Zerr I, Kretzschmar HA, de Pedro-Cuesta J, Calero-Lara M, Glatzel M, Aguzzi A, Bishop M, Knight R, Belay G, Will R, Mitrova E (2005) Genetic prion disease: the EUROCJD experience. Hum Genet 118:166–174
Kovacs GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS, Budka H (2002) Mutations of the prion protein gene phenotypic spectrum. J Neurol 249:1567–1582
Kulczycki J, Collinge J, Lojkowska W, Parnowski T, Wierzba-Bobrowicz T (2001) Report on the first polish case of the Gerstmann–Straussler–Scheinker syndrome. Folia Neuropathol 39:27–31
Liberski PP, Budka H (2004) Gerstmann–Straussler–Scheinker disease. I. Human diseases. Folia neuropathologica/Association of Polish Neuropathologists and Medical Research Centre. Polish Academy of Sciences 42(Suppl B):120–140
Lin KN, Wang PN, Liu CY, Chen WT, Lee YC, Liu HC (2002) Cutoff scores of the cognitive abilities screening instrument, Chinese version in screening of dementia. Dement Geriatr Cogn Disord 14:176–182
Majtenyi C, Brown P, Cervenakova L, Goldfarb LG, Tateishi J (2000) A three-sister sibship of Gerstmann–Straussler–Scheinker disease with a CJD phenotype. Neurology 54:2133–2137
Mallucci GR, Campbell TA, Dickinson A, Beck J, Holt M, Plant G, de Pauw KW, Hakin RN, Clarke CE, Howell S, Davies-Jones GA, Lawden M, Smith CM, Ince P, Ironside JW, Bridges LR, Dean A, Weeks I, Collinge J (1999) Inherited prion disease with an alanine to valine mutation at codon 117 in the prion protein gene. Brain 122(Pt 10):1823–1837
Masters CL, Gajdusek DC, Gibbs CJ Jr (1981) Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Straussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus-induced spongiform encephalopathies. Brain 104:559–588
Mastrianni JA (1998) The prion diseases: Creutzfeldt-Jakob, Gerstmann–Straussler–Scheinker, and related disorders. J Geriatr Psychiatry Neurol 11:78–97
Nai-Wen Guo H-CL, Wong Pai-Fang, Liao Kwong-Kun, Yan Sui-Hing, Lin Kong-Ping, Chang Cho-Yu, Hsu Tao-Chang (1988) Chinese Version and Norms of the Mini-Mental State Examination. Journal of Rehabilitation Medicine Association 16:52–59
Panegyres PK, Toufexis K, Kakulas BA, Cernevakova L, Brown P, Ghetti B, Piccardo P, Dlouhy SR (2001) A new PRNP mutation (G131 V) associated with Gerstmann–Straussler–Scheinker disease. Arch Neurol 58:1899–1902
Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, Hainfellner J, Reyes PF, Golden GT, Hauw JJ, Gajdusek DC, Gambetti P (1998) Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann–Straussler–Scheinker disease. Proc Natl Acad Sci USA 95:8322–8327
Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H (1999) Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233
Piccardo P, Dlouhy SR, Lievens PM, Young K, Bird TD, Nochlin D, Dickson DW, Vinters HV, Zimmerman TR, Mackenzie IR, Kish SJ, Ang LC, De Carli C, Pocchiari M, Brown P, Gibbs CJ Jr, Gajdusek DC, Bugiani O, Ironside J, Tagliavini F, Ghetti B (1998) Phenotypic variability of Gerstmann–Straussler–Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol 57:979–988
Prusiner SB (1998) Prions. Proc Natl Acad Sci U S A 95:13363–13383
Schmitz-Hubsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, Giunti P, Globas C, Infante J, Kang JS, Kremer B, Mariotti C, Melegh B, Pandolfo M, Rakowicz M, Ribai P, Rola R, Schols L, Szymanski S, van de Warrenburg BP, Durr A, Klockgether T, Fancellu R (2006) Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 66:1717–1720
Tanaka Y, Minematsu K, Moriyasu H, Yamaguchi T, Yutani C, Kitamoto T, Furukawa H (1997) A Japanese family with a variant of Gerstmann–Straussler–Scheinker disease. J Neurol Neurosurg Psychiatry 62:454–457
Teng EL, Hasegawa K, Homma A, Imai Y, Larson E, Graves A, Sugimoto K, Yamaguchi T, Sasaki H, Chiu D et al (1994) The Cognitive Abilities Screening Instrument (CASI): a practical test for cross-cultural epidemiological studies of dementia. Int Psychogeriatr 6:45–58 discussion 62
Wadsworth JD, Hill AF, Beck JA, Collinge J (2003) Molecular and clinical classification of human prion disease. Br Med Bull 66:241–254
Wadsworth JD, Jackson GS, Hill AF, Collinge J (1999) Molecular biology of prion propagation. Curr Opin Genet Dev 9:338–345
Wadsworth JD, Joiner S, Linehan JM, Cooper S, Powell C, Mallinson G, Buckell J, Gowland I, Asante EA, Budka H, Brandner S, Collinge J (2006) Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease-resistant wild-type and mutant prion protein. Brain 129:1557–1569
Wang PS, Wu YT, Hung CI, Kwan SY, Teng S, Soong BW (2008) Early detection of periodic sharp wave complexes on EEG by independent component analysis in patients with Creutzfeldt-Jakob disease. J Clin Neurophysiol 25:25–31
Wang Y, Qiao XY, Zhao CB, Gao X, Yao ZW, Qi L, Lu CZ (2006) Report on the first Chinese family with Gerstmann–Straussler–Scheinker disease manifesting the codon 102 mutation in the prion protein gene. Neuropathology 26:429–432
Yamada M, Tomimitsu H, Yokota T, Tomi H, Sunohara N, Mukoyama M, Itoh Y, Suematsu N, Otomo E, Okeda R, Matsushita M, Mizusawa H (1999) Involvement of the spinal posterior horn in Gerstmann–Straussler–Scheinker disease (PrP P102L). Neurology 52:260–265
Young K, Jones CK, Piccardo P, Lazzarini A, Golbe LI, Zimmerman TR Jr, Dickson DW, McLachlan DC, St George-Hyslop P, Lennox A et al (1995) Gerstmann–Straussler–Scheinker disease with mutation at codon 102 and methionine at codon 129 of PRNP in previously unreported patients. Neurology 45:1127–1134
Acknowledgments
This research was funded by Taipei Veterans General Hospital, Taiwan (V96C1-025 and V97C1-069), and the National Science Council, ROC (NSC96-2314-B-010-036-MY3 and NSC97-2314-B-010-036-MY3).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chi, NF., Lee, YC., Lu, YC. et al. Transmissible spongiform encephalopathies with P102L mutation of PRNP manifesting different phenotypes: clinical, neuroimaging, and electrophysiological studies in Chinese kindred in Taiwan. J Neurol 257, 191–197 (2010). https://doi.org/10.1007/s00415-009-5290-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-009-5290-4