
Abstract
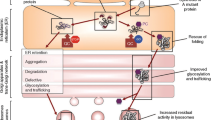
Enzyme replacement therapy (ERT) of the Anderson–Fabry disease (AFD) has changed the outcome of patients. However, ERT has some limitations: a restricted volume of distribution, requirement for intravenous access, and stimulation of the production of anti-drug antibodies. Studies of new drugs aiming to improve the clinical effectiveness and convenience of therapy have been reported. Migalastat, a pharmacological chaperone, increases available enzymate activity in patients with mutations amenable to the therapy, is now available for clinical practice. It is orally administered, and while clinical trial results are promising, long term real world follow up is awaited. PEGylated enzyme has a longer half-life and potentially reduced antigenicity, compared with standard preparations; investigation of whether a longer dosing interval is viable is under way. Moss-derived enzyme has a higher affinity for mannose receptors, and appears to have access to renal tissue. Substrate reduction therapy is based on reducing the catabolism processes of the glycosphingolipids, and is currently under investigation as monotherapy. Gene therapy has now been initiated in clinical trail of in vivo and ex vivo technologies with early results are emerging. ERT represents a certain milestone of therapy for AFD with Migalastat now a newly available option. Other agents in clinical trial prevent further potential opportunities to improve outcomes in AFD
Similar content being viewed by others

References
Germain PD (2010) Fabry disease. Orphan J Rare Dis 5:30
Arends M, Wanner C, Hughes D et al (2017) Characterization of classical and non-classical Fabry disease. J Am Soc Nephrol 28:1631–1641
Echevarria L, Benistan K, Toussaint A et al (2016) X-chromosome inactivation in female patients with Fabry disease. Clin Genet 89(1):44–54
Linthorst GE, Bouwman MG, Wijburg FA et al (2010) Screening for Fabry disease in high-risk populations: a systematic review. J Med Genet 47(4):217–222
Germain DP, Charrow J, Desnick RJ (2015) Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet 52(5):353–358
Beck M, Hughes D, Kampmann C et al (2017) Long-term outcomes with agalsidasealfa enzyme replacement therapy; Analysis using deconstructed composite events. Mol Genet Metab Rep 14:31–35
Lidove O, Wets M, Pintos-Morell G (2010) Effects of enzyme replacement therapy in Fabry disease—a comprehensive review of the medical literature. Genet Med 12(11):668–679
Lenders M, Brand E (2018) Effects of enzyme replacement therapy and antidrug antibodies inn patients with Fabry disease. J Am soc Nephrol 29:2265–2278
Medicine Agency Migalastat ( Galafold) :EU Summary of product characteristics 2018. https ://www.ema.europ a.eu/
Yam GH, Zuber C, Roth J et al (2005) A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding. Faseb J 19(1):12–18
Benjamin ER, Della Valle MC, Wu X et al (2017) The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med 19(4):430–438
Novak A, Huynh-Do U, Krayenbuehl P et al (2019) Fabry disease genotype, phenotype and Migalastat amenability: insights from a national cohort. J Inherit Metab Dis. https://doi.org/10.1002/jimd.12167
McCafferty EH, Scott LJ (2019) Migalastat A review in Fabry disease. Drugs 79(5):543–554
Amicus Therapeutics. Galafold™ (migalastat) capsules: NDA approval letter. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/20862 3Orig1s000ltr.pdf
Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR et al (2016) Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N Engl J Med 375(6):545–555
Hughes DA, Nicholls K, Shankar SP, Sunder-Plassmann G, Koeller D, Nedd K et al (2017) Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet 54(4):288–296
Levey AS, Stevens LA, Schmud CH et al (2009) A new equation to estimate glomerular filtration rate. Ann Inter Med 150:604–612
Muntze J, Gensler D, Maniuc O et al (2019) Oral chaperone therapy: Migalastat for treating Fabry disease: enzymatic response and serum biomarker changes after 1 year. Clin Pharmacol Ther 105(5):1224–1233
Lenders M, Stappers F, Niemietz C et al (2019) Mutation-specific Fabry disease patient-derived cel model to evaluate the amenability to chaperone therapy. J Med Genet 56(8):548–556
Germain DP, Nicholls K, Giugliani R et al (2019) Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat-amenable variants : data from the phase 3 randomised, multicentre, double-blind clinical trial and extension study. Genet Med 21(9):1987–1997
Veronese F, Pasut G (2005) PEGylation, successful approach to drug delivery. Drug Discov Today 10(21):1451–1458
Tekoah Y, Shulman A, Kizhner T et al (2015) Large-scale production of pharmaceutical proteins in plant cell culture-the Protalix experience. Plant Biotechnol J 13(8):1199–1208
Kizhner T, Azulav Y, Hainrichson M et al (2015) Characterizarion of a chemically modified plant cell culture expressed human α-Galactosidase-A enzyme for treatment of Faby disease. Mol Genet Met 114:259–267
Shiffmann R, Goker-Alpan O et al (2019) Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease provides sustained plasma concentrations and favourable pharmacodynamics: a 1 year Phase 1/2 clinical trial. J Inherit Metab Dis 42(3):534–544
Ruderfer I, Shulman A, Kizhner A, Azulav Y, Natal Y, Tokoa Y, Shaaltiel Y (2018) Development and analytical characterization of Pegunigalsidase alfa, a chemical crosslinked plant recombinant human α-galactosidase A for treatment of Fabry disease. Bioconjug Chem 29:1640–1648
Hughes D, Giraldo P, Holida M et al (2018) Pegunigalsidas alfa a novel PEGylated ERT for Fabry disease: two-year safety and efficacy follow-up. Mol Genet Metab 123:S15–S153
Warnock D, Eric W, Shiffmann R et al (2018) Analysis of the baseline characteristics of Fabry disease patients screened for Pegunigalsidase alfa phase III BALANCE study. Mol Genet Metab 126:S150–S152
Holida MD, Bernat J, Longo N et al (2019) Once every 4 weeks-2mg/kgb of Pegunigalsidase alfa for treating Fabry disease Preliminary results of phase 3. Mol Genet Metab 126:S17–S156
Linhart A, Nicholls M, West M et al (2019) Pegunigalsidase alfa for treatment of fabry disease: preliminary results from a phase III open label; switch over study from agalsidase alfa. Mol Genet Metab 126:S17–S156
Koiprova A, Stemmer C, Altmann F et al (2002) Targeted knockouts of Physcomitrella lacking plant-specific immunogenic N-glycans. Plant Biotechnol J 2:517–523
Shen J-S, Busch A, Day TS et al (2016) Mannose receptor-mediated delivery of moss-made α-galactosidase A efficiently corrects enzyme deficiency in Fabry mice. J Inherit Metab Dis 39:293–303
Hennermann J, Arash-Kaps L, Feke G et al (2019) Pharmacokinetics, pharmacodynamics and safety of moss-a Galactosidase A in patients with Fabry disease. J Inherit Metab Dis 42:1–7
Shiffmann R, Murray GJ, Treco D et al (2000) Infusion of α-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci 97:365–370
Guerard N, Oder D, Nordbeck P et al (2018) Lucerastat, an iminosugar for substrate reduction: tolerability, pharmacodynamics, and pharmacokinetics in patients with Fabry disease on enzyme replacement. Clin Pharmacol Ther 103:703–711
Platt FM, Jayakumar M (2008) Substrate reduction therapy. Acta Pediatr 97:88–93
Ashe KM et al (2015) Efficacy of enzyme substrate reduction therapy with a novel antagonist of glucosylceramide synthase for Fabry disease. Mol Med 21:389–399
Marshall R et al (2010) Substrate reduction augments the efficacy of enzyme therapy in a mouse model of Fabry disease. Plos One 5:5033
Baek RC, Kasperzyk JL, Platt FM et al (2008) Butyldeoxgalactonojirimycin reduces brain ganglioside and GM2 content in neonatal Sandhoff disease mice. Neurochem Int 52(6):1125–1133
Weford RWD et al (2017) Lucerastat, an iminosugar substrate reduction therapy for Fabry disease: preclinical evidence. World LDN 2017 Abs 360
Guerard N, Zwingelstein C, Dingemanse J (2017) Lucerastat, an iminosugar for substrate reduction therapy: tolerability and safety in subjects with mild, moderate and severe renal impairment. J Clin Pharmacol 57:1425–1431
Guerard N, Oder D, Norbeck P et al (2018) Lucerastat an iminosugar for substrate reduction therapy: tolerability, pharmacodynamics, and pharmacokinetics in patients with Fabry disease on enzyme replacement. Clin Phatmacol Ther 103(4):703–711
Arends M, Wijburg FA, Wanner C et al (2017) Favourable effect of early versus late enzyme replacement therapy on plasma globotriaosylsphingosine in men with classical Fabry disease. Mol Genet Metab. https://doi.org/10.1016/j.ymgme.2017.05.001
Simonetta I, Tuttolomondo A, Di Chiara T et al (2018) Genetics and gene therapy of Anderson-Fabry disease. Curr Gene Ther 18:96–106
Murtaza S, Nagree MS, Scalia S et al (2019) An update on gene therapy for lysosomal storage disorders. Exp Opin Biol Ther 19:655–670
de Garibay APR, Solinis MA, Rodriguez-Gascon A (2013) Gene therapy for Fabry disease: a review of the Literature. BioDrugs 27:237–246
Nagree MS, Faber ML, Tate E et al (2019) In vivo enrichment of traduced cells to enhance gene therapy. MGM 126(S17):S156
Jeyakumar J, Kia A, McIntosh J et al (2019) Liver direct therapy corrects Fabry disease in mice. MGM 126:S17–S156
Huston MW, Yasuda M, Pagant S et al (2019) Liver-targeted AAV gene therapy vectors produced by a clinical scale manufacturing process result in high, continuous therapeutic level of enzyme activity and effective substrate reduction model Fabry disease. MGM 126:2
Yoshimitsu M, Tao K, Fan X et al (2019) Neonatal gene therapy with a lentiviral vector results in therapeutic levels of α galactosidase A Correction in Fabry mice. MGM 126:S17–S156
Saida O, Biferi MG, San Millan B et al (2019) Functional evaluation of an AAV (based vector expressing alpha/galactosidase A for potential gene therapy. Mol Genet 126:S17–S1562
Kevany B, Kerns S, Padegimas L, Miller J (2019) AAV gene therapy for treatment of Fabry disease A novel capsid with improved tropism to heart, kidney and CNS and improved GLA expression. MGM 126:S17–S156
Medin JA, Khan A, Huang J et al (2019) FACTs Fabry gene clinical trial: two-year data. MGM 126:S17–S156
Takahashi H, Hirai Y, Migita M et al (2002) Long-term systemic therapy of Fabry disease in a knockout mouse by adeno-associated virus-mediated muscle-directed gene transfer. PNAS 99(21):13777–13782
Jung SC, Han IP, Limaye A et al (2001) Adeno-associated viral vector-mediated gene transfer results in long term enzymatic and functional correction in multiple organs of Fabry mice. PNAS 98:12676–12681
Ziegler RJ, Cherry M, Barbon CM et al (2007) Correction of the biochemical and functional deficits in Fabry mice following AAV8-mediated hepatic expression of alpha-galactosidase A. Mol Ther 15(3):492–500
Sabatino DE, Mackenzie TC, Peranteau W et al (2007) Persistent expression og hF.IX after tolerance induction by in utero or neonatal administration of AAV-1-F. IX in haemophilia B mice. Mol Ther 15(9):1677–1685
de Garibay APR, Delgado D, Del Porto-Rodriguez M et al (2012) Multicomponent as nonviral vectors for the treatment of Fabry disease by gene therapy. Drug Des Dev Ther 6:303–310
Lenders M, Neuber LP, Rudnicki M et al (2018) Replacement therapy on neutralizing antidrug antibodies titters and clinical outcome in patients with Fabry. J Am Soc Nephrol 29(12):2879–2889
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Sandro Feriozzi has received honoraria for advisory boards and travel expenses from Amicus, Sanofi Genzyme, and Takeda Shire*; received fees from Amicus, Otsuka, Sanofi Genzyme, and Takeda/Shire*. Deralynn Hughes has received travel and research grants and honoraria for speaking and advisory boards from Amicus, Genzyme, Protalix, and Shire.
Human participants and/or animals
This manuscript is a review of previously published papers and there are no new, unpublished data. All data were reported in official journals in which it is declared that the studies have been approved by the appropriate institutional and/or national research ethics committee and have been performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
This manuscript is a review of previously published papers and there are no new, unpublished data. In all papers it was reported that the informed consent of patients was obtained.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Feriozzi, S., Hughes, D.A. New drugs for the treatment of Anderson–Fabry disease. J Nephrol 34, 221–230 (2021). https://doi.org/10.1007/s40620-020-00721-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-020-00721-4