
Abstract
The antiviral agent remdesivir (Veklury®; Gilead Sciences), nucleotide analogue prodrug, has broad-spectrum activity against viruses from several families. Having demonstrated potent antiviral activity against coronaviruses in preclinical studies, remdesivir emerged as a candidate drug for the treatment of the novel coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, during the current global pandemic. Phase III evaluation of remdesivir in the treatment of COVID-19 commenced in early 2020 and has thus far yielded promising results. In late May 2020, Taiwan conditionally approved the use of remdesivir in patients with severe COVID-19. This was followed by a rapid succession of conditional approvals in various countries/regions including the EU and Canada. Preceding these conditional approvals, an emergency use authorization for remdesivir had been granted in the USA (on 1 May 2020) and a special approval for emergency use was granted in Japan (on 7 May 2020). This article summarizes the milestones in the development of remdesivir leading to its first conditional approval for the treatment of COVID-19.
Similar content being viewed by others
A nucleotide analogue prodrug is being developed by Gilead Sciences for the treatment of COVID-19 |
Received its first emergency use authorization on 1 May 2020 in the USA |
Received its first conditional approval in late May 2020 in Taiwan |
Approved for use in patients with severe COVID-19 |
1 Introduction
Remdesivir (Veklury®; Gilead Sciences), a prodrug of an adenosine nucleotide analogue, is an antiviral agent with broad-spectrum activity against viruses from several families [1,2,3,4]. Remdesivir was previously under development for the treatment of Ebola virus disease in the wake of the 2014-2016 Ebola outbreak in West Africa. While it was a promising therapeutic agent for Ebola virus disease in preclinical studies [4], monoclonal antibodies outperformed remdesivir in a phase III clinical trial [5] and remdesivir is no longer being developed in this indication. The antiviral activity of remdesivir against coronaviruses, however, has rendered the drug of great interest during the current global pandemic. The novel coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, was first reported in Wuhan, China, in December 2019 [6]. The World Health Organization declared COVID-19 a Public Health Emergency of International Concern on 30 January 2020 and a pandemic on 11 March 2020 [6], spurring an international effort to rapidly identify treatments that might ease the burden on healthcare systems.
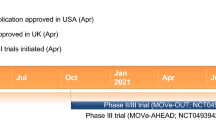
Phase III trials of remdesivir in COVID-19 were initiated as early as February 2020. Based on data from the multinational phase III ACTT-1 and SIMPLE-severe trials, remdesivir received an emergency use authorization in the USA on 1 May 2020 [7, 8] and a special approval for emergency use in Japan on 7 May 2020 [9, 10]. Remdesivir received its first conditional approval for use in patients with severe COVID-19 in Taiwan in late May 2020, with this conditional approval requiring the pharmaceutical company to implement a risk management plan to ensure safety [11].

Key milestones in the development of remdesivir for use in COVID-19. NDA New Drug Application
In June and July 2020, remdesivir was conditionally approved in several other countries/regions worldwide, including the EU [12], Singapore [13], Australia [14], South Korea [15] and Canada [16]. In the EU and Canada, remdesivir represents the first approved COVID-19 treatment [12, 16] and is indicated for the treatment of COVID-19 in adults and adolescents (aged ≥ 12 years and with a body weight ≥ 40 kg) with pneumonia requiring supplemental oxygen; the safety and efficacy of remdesivir in paediatric patients < 12 years of age and weighing < 40 kg has not been established [17, 18]. Also during June and July 2020, remdesivir was approved as an emergency medication in the UAE [19] and received restricted approval for emergency use in India (with several generic formulations of remdesivir also being approved, manufacturers of which include Cipla Ltd., Hetero Drugs, Mylan and Jubilant Pharma) [20,21,22]. On 10 August 2020, a New Drug Application for remdesivir for the treatment of COVID-19 was submitted to the US FDA [23].
Remdesivir is administered intravenously and is available as a solution and/or lyophilized powder for infusion over 30–120 min [7, 9, 17, 18]. The typical recommended dosage regimen is a single 200 mg loading dose on day 1 and 100 mg given once daily on day 2 onwards, for a total treatment duration of at least 5 days and no more than 10 days. Kidney and liver function should be determined prior to initiating treatment with remdesivir, as remdesivir should not be used in patients with an estimated glomerular filtration rate (eGFR) of < 30 mL/min or in patients with alanine aminotransferase (ALT) ≥ 5 times the upper limit of normal (ULN) [7, 9, 17, 18]. Some drugs are not suitable for coadministration with remdesivir (e.g. chloroquine phosphate, hydroxychloroquine sulphate) [7, 17, 18]; consult local prescribing information for further details.

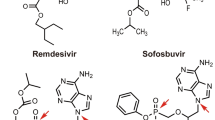
Chemical structure of remdesivir
The safety and efficacy of remdesivir (including in paediatric patients and in combination with anti-inflammatory agents) continues to be evaluated in ongoing clinical trials and compassionate use programmes. In July 2020, it was announced that an inhaled solution of remdesivir for the potential outpatient treatment of COVID-19 is undergoing phase I development [24].
1.1 Company Agreements
In December 2015, Gilead Sciences and Ligand Pharmaceuticals entered into a supply agreement [25]. Under its terms, Ligand Pharmaceuticals would supply Gilead with a uniquely modified cyclodextrin (Captisol®), the chemical structure of which was rationally designed to optimize the solubility and stability of drugs including remdesivir [26], for use in a Captisol-enabled program directed against Ebola virus disease (in addition to other programs, if expansions to the agreement were agreed by the companies) [25]. In turn, Ligand would receive an upfront Drug Master File reference fee, as well as commercial revenue from the shipment of Captisol to Gilead [25]. As of April 2020, Ligand is supplying Captisol to Gilead for use in clinical trials evaluating remdesivir in the treatment of COVID-19 [27].
In the first half of 2020, Gilead Sciences entered into non-exclusive voluntary licensing agreements with a number of generic pharmaceutical manufacturers based in India, Egypt and Pakistan (e.g. Mylan, Cipla Ltd., Hetero Labs Ltd., Dr. Reddy’s Laboratories Ltd., Zydus Cadila Healthcare Ltd., Eva Pharma, Ferozsons Laboratories, Jubilant Lifesciences and Syngene, a Biocon company) [28]. The licensing agreements serve to expand access to remdesivir for the treatment of COVID-19, through allowing the companies to manufacture and distribute remdesivir in 127 countries (mostly of low- or middle-income status). All licenses are royalty-free until the World Health Organization announces the end of the Public Health Emergency of International Concern regarding COVID-19 or until another pharmaceutical product or vaccine is approved for the treatment or prevention of COVID-19 (whichever is earlier) [28].
2 Scientific Summary
2.1 Pharmacodynamics
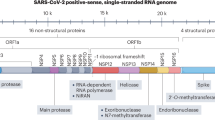
Remdesivir, a nucleotide analogue prodrug, is metabolized within host cells to form a pharmacologically active nucleoside triphosphate [29,30,31,32]. By acting as an adenosine triphosphate (ATP) analogue, remdesivir triphosphate competes with the natural ATP substrate for integration into nascent viral RNA chains by RNA-dependent RNA polymerase. Once remdesivir triphosphate has been mistakenly incorporated into the chain and a small number of subsequent nucleotides (typically three, in coronaviruses) have been added, RNA synthesis is terminated [29,30,31,32]. Remdesivir has broad-spectrum antiviral activity against numerous viruses including Ebola virus, Nipah virus and respiratory syncytial virus, as well as endemic and zoonotic coronaviruses [1,2,3,4]. In primary human airway epithelial cell cultures, remdesivir inhibited the replication of severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) with submicromolar half-maximum inhibitory concentration (IC50) values [3]. These results heralded remdesivir as an antiviral with potential activity against novel coronaviruses.
In vitro, remdesivir displayed antiviral activity against SARS-CoV-2 in primary human airway epithelial cultures, potently and dose-dependently inhibiting SARS-CoV-2 replication with a half-maximal effective concentration (EC50) of 0.01 μM [33]. This antiviral effect appears to be virus-specific; remdesivir is not cytotoxic at ≤ 10 μg in this culture system [3, 33]. In Vero E6 cells, EC50 values of remdesivir and its metabolite GS-441524 against SARS-CoV-2 were 1.65 μM and 0.47 μM, respectively, reflecting the lower capacity of Vero E6 cells to metabolize remdesivir [33]. When clinically relevant concentrations of remdesivir and chloroquine phosphate were co-incubated in HEp-2 cells infected with respiratory syncytial virus, chloroquine phosphate dose-dependently antagonized the antiviral activity of remdesivir. With increasing concentrations of chloroquine phosphate, higher remdesivir EC50 values and reduced remdesivir triphosphate formation in normal human bronchial epithelial cells were observed [18]. Consequently, the coadministration of remdesivir and chloroquine phosphate or hydroxychloroquine sulphate is not recommended [17, 18].
In mice infected with a chimeric SARS-CoV virus encoding the RNA-dependent RNA polymerase of SARS-CoV-2, treatment with remdesivir significantly (p = 0.0012 vs vehicle) reduced lung viral load and significantly (p ≤ 0.004) ameliorated the loss of pulmonary function seen in vehicle-treated mice [33]. Similar therapeutic effects were observed in a rhesus macaque model of SARS-CoV-2 infection [34]. Compared with macaques receiving vehicle solution, macaques treated with remdesivir had significantly (p ≤ 0.0069) less severe pulmonary infiltration on radiographs and significantly (p ≤ 0.0004) reduced clinical scores. Following euthanasia, both viral load and lung area affected by lesions were significantly (p ≤ 0.0002 vs vehicle) reduced in the lungs from macaques treated with remdesivir [34].
Although the potential for QT prolongation has not been thoroughly evaluated in humans, current non-clinical and clinical data do not indicate any risk of QT prolongation with remdesivir [17, 18, 35].
2.2 Pharmacokinetics
Exposures of remdesivir and its metabolites increased in a dose-proportional manner across the evaluated dose range (3–225 mg) in healthy adults following intravenous administration [36]. Solution and lyophilized formulations of remdesivir had comparable pharmacokinetic parameters [36]. In healthy adults intravenously administered the adult dosage regimen of remdesivir, plasma concentrations of remdesivir peaked at the end of infusion (irrespective of dose level) and then rapidly declined [18]. Plasma concentrations of GS-441524, the predominant circulating metabolite, peaked 1.5–2.0 h after the start of a 30 min infusion. Remdesivir and GS-441524 have human plasma protein binding rates of ≈ 88% and 2%, respectively [18].
Remdesivir is highly metabolized, undergoing metabolic activation to intracellularly form GS-443902, the pharmacologically active nucleoside analogue triphosphate [18, 36]. Initially, remdesivir undergoes hydrolysis by esterases and forms GS-704277, the intermediate metabolite. Phosphoramidate cleavage of GS-704277 and further phosphorylation of the resultant nucleoside analogue monophosphate produces the nucleoside triphosphate GS-443902, while dephosphorylation of phosphorylated metabolites can produce the nucleoside analogue GS-441524 [18, 36].
The main elimination pathway for GS-441524 is renal clearance [18]. Following intravenous administration of a single 150 mg radiolabelled dose of remdesivir, ≈ 74% and 18% of the dose was recovered in urine and feces, respectively. A large proportion of the dose recovered in urine was GS-441524 (49%); only 10% was remdesivir. Remdesivir and GS-441524 had median terminal half-lives of ≈ 1 and 27 h, respectively [18].
It is unknown whether age, sex, race or hepatic impairment impact upon the pharmacokinetic properties of remdesivir [18]. Although the pharmacokinetics of remdesivir and GS-441524 have not been evaluated in patients with renal impairment, plasma levels of GS-441524 may theoretically be increased in patients with decreased renal function [18]; all patients should have their eGFR determined prior to commencing treatment with remdesivir and during treatment as clinically appropriate [17, 18]. Remdesivir should not be used in patients with an eGFR of < 30 mL/min [17, 18].
In vitro, remdesivir is a substrate for CYP2C8, 2D6 and 3A4, P-gp and OATP1B1, as well as esterases in tissue and plasma [18, 35, 37]. Strong inhibitors of the hydrolytic pathway or of CYP2C8, 2D6 or 3A4 may increase remdesivir exposure [18]; the use of strong inducers (e.g. rifampicin) may decrease remdesivir plasma concentrations and is therefore not recommended [17, 18]. Dexamethasone, a moderate inducer of CYP3A and P-gp, is unlikely to have clinically significant interactions with remdesivir [18]. Remdesivir is an inhibitor of CYP3A4, OATP1B1 and OATP1B3 in vitro [18, 35, 37]. While no data are available, plasma concentrations of drugs that are CYP3A or OATP1B1/1B3 substrates may be transiently increased by remdesivir and it is thus suggested that CYP3A4 or OATP1B1/1B2 substrates should be administered ≥ 2 h after remdesivir [17, 18]. As remdesivir is an inducer of CYP1A2 and possibly CYP3A in vitro, CYP1A2 or CYP3A4 substrates with narrow therapeutic indices may suffer a loss of efficacy if coadministered with remdesivir [18]. Consult local prescribing information for further details concerning potential drug interactions.
2.3 Therapeutic Efficacy
2.3.1 In Clinical Trials
In the randomized, open-label, multinational, phase III SIMPLE-moderate trial (NCT04292730), patients with moderate COVID-19 pneumonia receiving 5-day remdesivir treatment were significantly more likely to have clinical improvement at day 11 than those receiving standard of care (SOC) alone (odds ratio (OR) 1.65; 95% CI 1.09–2.48; p = 0.017); the difference between patients receiving 10-day remdesivir and those receiving SOC alone did not reach statistical significance (OR 1.31; 95% CI 0.88–1.95) [38]. At day 11, ≥ 2-point improvements on a 7-point ordinal scale were achieved by 70% and 65% of patients in the 5-day and 10-day remdesivir groups (vs 61% of patients in the SOC group) and ≥ 1-point improvements were achieved by 76% and 70% (vs 66%). Deaths occurred in two patients receiving 10-day remdesivir (1%) and four patients receiving SOC alone (2%). SIMPLE-moderate enrolled hospitalized adults and adolescents ≥ 12 years of age with confirmed SARS-CoV-2 infection and evidence of pneumonia without reduced oxygen levels. Patients received intravenous remdesivir (200 mg on day 1 and 100 mg on subsequent days) plus SOC for 5 days (n = 191) or 10 days (n = 193), or SOC alone (n = 200) [38].
In the randomized, open-label, multinational, phase III SIMPLE-severe trial (NCT04292899) in patients with severe COVID-19, there was no significant difference between 5-day and 10-day remdesivir recipients with respect to distribution of clinical status (assessed on a seven-point ordinal scale) at day 14 (p = 0.14; primary outcome) after adjusting for baseline clinical status [39]. At day 14, a clinical improvement of ≥ 2 points on the ordinal scale was achieved by 64.5% of 5-day remdesivir recipients and 54.3% of 10-day remdesivir recipients (baseline-adjusted treatment difference − 6.5%; 95% CI − 15.7 to 2.8). SIMPLE-severe enrolled adults and adolescents ≥ 12 years of age with confirmed SARS-CoV-2 infection, radiographic evidence of pulmonary infiltrates and either an oxygen saturation of ≤ 94% while breathing ambient air or receiving supplemental oxygen. The trial excluded patients receiving mechanical ventilation and extracorporeal membrane oxygenation (ECMO) at screening. Patients were randomized to receive intravenous remdesivir (200 mg on day 1 and 100 mg once daily on subsequent days) for either 5 days (n = 200 treated) or 10 days (n = 197), in addition to continued SOC. At baseline, clinical status was significantly poorer in the 10-day remdesivir group than in the 5-day group (p = 0.02) [39].
In a robust comparison of interim data from SIMPLE-severe and a concurrent retrospective cohort study, remdesivir was associated with a significantly improved recovery rate relative to SOC alone at day 14 (74.4% vs 59.0%; adjusted OR 2.03; 95% CI 1.34–3.08; p < 0.001) [40]. In addition, remdesivir significantly reduced mortality relative to SOC alone (7.6% vs 12.5%; OR 0.38; 95% CI 0.22–0.68; p = 0.001). At day 14, significantly (p ≤ 0.01) more remdesivir recipients than SOC recipients achieved a ≥ 1-point or ≥ 2-point improvement in clinical status on an ordinal scale. The preplanned comparative analysis included adults hospitalized with severe COVID-19 receiving intravenous remdesivir plus SOC in SIMPLE-severe (n = 312) or SOC alone in the real-world cohort (n = 818). Groups were generally well balanced in terms of baseline factors, with propensity score methods used to approximate a randomized controlled trial [40].
Based on preliminary results from the randomized, double-blind, placebo-controlled, multinational phase III ACTT-1 trial (NCT04280705) in patients with COVID-19, remdesivir significantly reduced time to recovery relative to placebo (median 11 days vs 15 days; rate ratio for recovery 1.32; 95% CI 1.12–1.55; p < 0.001) [primary endpoint] [41]. In patients with severe disease, time to recovery was 12 days with remdesivir and 18 days with placebo (rate ratio 1.37; 95% CI 1.15–1.63); there was no significant treatment difference for patients with mild/moderate disease (time to recovery 5 days in each group) [18]. Patients randomized during the first 10 days after symptom onset had a recovery rate ratio of 1.28 (95% CI 1.05–1.57), while patients randomized more than 10 days after symptom onset had a recovery rate ratio of 1.38 (95% CI 1.05–1.81) [41]. The odds of improvement (based on an eight-point ordinal scale) were significantly higher with remdesivir than with placebo at day 15 (OR 1.50; 95% CI 1.18–1.91; p = 0.001) [key secondary endpoint], while mortality rate did not significantly differ between the groups (HR for death 0.70; 95% CI 0.47–1.04). Kaplan-Meier estimates of mortality by 14 days were 7.1% and 11.9% with remdesivir and placebo, respectively. ACTT-1 enrolled adults hospitalized with COVID-19 and evidence of lower respiratory tract involvement. Patients received either intravenous remdesivir (200 mg on day 1 followed by 100 mg daily for the next ≤ 9 days; n = 538 analyzed) or matching placebo (n = 521). There was a median of 9 days between symptom onset and randomization, and the majority of patients (88.7%) had severe disease at enrolment. Recovery was defined as either discharge from hospital or hospitalization for infection-control reasons only. On the basis of the preliminary finding of reduced recovery time with remdesivir versus placebo, early unblinding of results was recommended by the data and safety monitoring board [41].
In a randomized, double-blind, placebo-controlled, multicentre phase III trial (NCT04257656) conducted in Wuhan, China, remdesivir did not significantly reduce time to clinical improvement relative to placebo (median 21 days vs 23 days; HR 1.23; 95% CI 0.87–1.75) [intention-to-treat population; primary endpoint] in patients with severe COVID-19 [42]. A similar result was observed in the per protocol population. In patients receiving remdesivir or placebo within 10 days of symptom onset, time to clinical improvement was 18 days with remdesivir versus 23 days with placebo (HR 1.52; 95% CI 0.95–2.43) in the intention-to-treat population. Mortality at day 28 was 14% in the remdesivir group and 13% in the placebo group (difference 1.1%; 95% CI −8.1 to 10.3). This trial enrolled adults admitted to hospital with laboratory-confirmed SARS-CoV-2 infection, pneumonia confirmed by chest imaging and an oxygen saturation of ≤ 94% on room air or an arterial oxygen partial pressure to fractional inspired oxygen ratio of ≤ 300 mm Hg. Patients were within 12 days of symptom onset at enrolment. Patients were randomized to either intravenous remdesivir (single daily infusions of 200 mg on day 1 and 100 mg on days 2–10; n = 158 enrolled) or volume-matched placebo infusions (n = 79). Clinical improvement was defined as a two-point reduction on a six-point ordinal scale of clinical status or live discharge from hospital (whichever was earlier) within 28 days of randomization. It should be noted that this study was underpowered (statistical power reduced from 80% to 58%), not reaching its target enrolment due to control of the outbreak in the region [42].
2.3.2 In Compassionate Use
In a multinational sample of patients with COVID-19 who received remdesivir in the compassionate use program (n = 163), the rate of clinical improvement was 47% after a median follow-up of 15 days [43]. A ≥ 2-point clinical improvement was achieved by 41% of patients and 30% of patients were discharged from hospital. The overall mortality rate was 20%. At baseline, 64% of patients were receiving invasive oxygen support [43].
In an earlier analysis of data from adults with COVID-19 treated with compassionate use remdesivir (n = 53), the overall rate of clinical improvement was 68% after a median follow-up of 18 days [44]. The discharge rate was 47% and the mortality rate was 13%. At baseline, 57% of patients were receiving mechanical ventilation and 8% were receiving ECMO [44].
Among pregnant women (n = 67) and postpartum women (n = 19) who received compassionate use remdesivir for severe COVID-19, rates of clinical improvement were 96% and 89%, respectively, at day 28 [45]. For pregnant women, the recovery rate was 93% and the discharge rate was 90%; for postpartum women, the respective rates were 89% and 84%. At baseline, 95% of postpartum women and 40% of pregnant women were on mechanical ventilation or ECMO. Postpartum women were those who delivered prior to receiving their first dose of remdesivir [45].
In paediatric patients (aged 0–17 years) with severe COVID-19 treated with compassionate use remdesivir (n = 77), the clinical improvement rate was 88% at day 28 [46]. Clinical recovery occurred in 80% of patients on ventilators/ECMO at baseline and in 87% of patients not requiring invasive oxygen support. At baseline, 51% of patients were receiving invasive mechanical ventilation or ECMO [46].
Patients receiving remdesivir via the compassionate use program and included in the aforementioned analyses had confirmed SARS-CoV-2 infection and were receiving oxygen support or had an oxygen saturation of ≤ 94% while breathing ambient air [43,44,45,46]. They received intravenous remdesivir for up to 10 days; adult patients and paediatric patients weighing ≥ 40 kg received 200 mg on day 1 and 100 mg daily on each remaining day [43,44,45,46], while paediatric patients weighing < 40 kg received 5 mg/kg on day 1 and 2.5 mg/kg on each remaining day [46].
2.4 Adverse Events
Remdesivir was generally well tolerated when administered intravenously in clinical trials [38, 39, 41, 42] and the compassionate use program [43,44,45,46]. In the ACTT-1 safety population (n = 541 and 522 patients treated with remdesivir and placebo, respectively, for ≤ 10 days), the most common adverse events (AEs) reported in remdesivir recipients were anemia or decreased hemoglobin (7.9% vs 9.0% of placebo recipients); acute kidney injury, decreased eGFR or creatinine clearance, or increased blood creatinine (7.4% vs 7.3%); pyrexia (5.0% vs 3.3%); hyperglycemia or increased blood glucose level (4.1% vs 3.3%); and increased ALT and/or aspartate aminotransferase (AST) [4.1% vs 5.9%] [41]. Grade 3 or 4 AEs occurred in 28.8% of remdesivir recipients and 33.0% of placebo recipients, while serious AEs occurred in 21.1% and 27.0%. Two serious AEs in each group were considered to be related to treatment. Serious respiratory failure AEs occurred in 5.2% of remdesivir recipients and 8.0% of placebo recipients. No deaths were judged by site investigators to be related to treatment assignment [41].
In SIMPLE-severe (n = 200 and 197 receiving 5-day and 10-day remdesivir, respectively) [39] and SIMPLE-moderate (n = 191, 193 and 200 receiving 5-day remdesivir, 10-day remdesivir and SOC alone, respectively) [38], AE rates were similar between patients treated with 5-day remdesivir (70% in SIMPLE-severe and 51% in SIMPLE-moderate) and patients treated with 10-day remdesivir (74% and 55%, respectively). In SIMPLE-moderate, the corresponding rate in patients receiving SOC alone was 45% [38]. In both trials, the most common AE was nausea (10% of 5-day remdesivir recipients and 9% of 10-day remdesivir recipients in each study; 3% of patients receiving SOC alone in SIMPLE-moderate) [38, 39]. Grade ≥ 3 AEs occurred in 30% and 43% of patients in the 5-day and 10-day remdesivir groups, respectively, in SIMPLE-severe [39] and in 10% and 11% of patients in the respective groups in SIMPLE-moderate (vs 12% of patients receiving SOC alone) [38]. Serious AEs occurred in 21% and 35% of patients in the 5-day and 10-day remdesivir groups, respectively, in SIMPLE-severe [39] and in 4% of patients in each remdesivir group in SIMPLE-moderate (vs 9% of patients receiving SOC alone) [38]. With respect to laboratory abnormalities, grade ≥ 3 (≥ 5 × ULN) increases in ALT and AST each occurred in 7% of remdesivir recipients in SIMPLE-severe and in 3% and 2% of remdesivir recipients (vs 7% and 6% of SOC recipients) in SIMPLE-moderate [18]. In SIMPLE-severe, AEs led to treatment discontinuation in 4% and 10% of patients in the 5-day and 10-day remdesivir groups, respectively [39].
Given that transaminase elevations have been observed with remdesivir in clinical trials, liver function should be evaluated prior to initiation of remdesivir and monitored during treatment as clinically appropriate [18]. Due to a lack of data, remdesivir should only be used in patients with hepatic impairment if the potential benefit outweighs the potential risk. In patients with ALT ≥ 5 × ULN at baseline, remdesivir should not be initiated. Remdesivir should be discontinued if a patient develops ALT ≥ 5 × ULN (and may be restarted when ALT < 5 × ULN) or ALT elevation in conjunction with signs/symptoms of liver inflammation or increasing conjugated bilirubin, alkaline phosphatase or international normalized ratio during treatment [18].
Hypersensitivity reactions (including infusion-related reactions and anaphylactic reactions) can occur with remdesivir [18]. Clinicians can consider slower infusion rates (≤ 120 min maximum infusion time) to potentially prevent signs and symptoms of these. If a clinically significant hypersensitivity reaction occurs, remdesivir administration should be immediately discontinued and appropriate treatment initiated [18].
Compassionate use remdesivir was generally well tolerated in pregnant or postpartum women with severe COVID-19; no new safety signals were identified [45]. Due to limited data, however, remdesivir should be avoided during pregnancy unless required based on the clinical condition of the woman [18]. Patients of child-bearing potential should use effective contraception during treatment with remdesivir. Breast-feeding patients should choose between discontinuing breastfeeding and discontinuing/abstaining from treatment with remdesivir [18].
2.5 Ongoing Clinical Trials
Ongoing multinational clinical trials include the adaptive, randomized, double-blind, phase III ACTT-2 trial (NCT04401579), which will evaluate the combined use of remdesivir and baricitinib versus remdesivir plus placebo in hospitalized adults with COVID-19, and the randomized, double-blind, phase III REMDACTA trial (NCT04409262), which will compare remdesivir plus tocilizumab with remdesivir plus placebo in the treatment of hospitalized adults and adolescents ≥ 12 years of age with COVID-19 pneumonia. REMDACTA is currently recruiting. Also currently recruiting is the single-arm, open-label phase II/III CARAVAN trial (NCT04431453) to evaluate the safety, tolerability, pharmacokinetics and efficacy of remdesivir in paediatric patients (aged from birth to < 18 years) with COVID-19.
3 Current Status
Remdesivir received its first conditional approval in late May 2020 in Taiwan for use in patients with severe COVID-19.
References
Brown AJ, Won JJ, Graham RL, et al. Broad spectrum antiviral remdesivir inhibits human endemic and zoonotic deltacoronaviruses with a highly divergent RNA dependent RNA polymerase. Antiviral Res. 2019;169:104541.
Lo MK, Jordan R, Arvey A, et al. GS-5734 and its parent nucleoside analog inhibit filo-, pneumo-, and paramyxoviruses. Sci Rep. 2017;7:43395.
Sheahan TP, Sims AC, Graham RL, et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med. 2017;9:396.
Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. 2016;531(7594):381–5.
Mulangu S, Dodd LE, Davey RT, et al. A randomized, controlled trial of Ebola virus disease therapeutics. N Engl J Med. 2019;381(24):2293–303.
World Health Organization. Timeline of WHO’s response to COVID-19. 2020. http://www.who.int/. Accessed 24 Jul 2020.
Gilead Sciences. Fact sheet for health care providers emergency use authorization (EUA) of remdesivir (GS-5734™), and full EUA prescribing information. 2020. http://www.fda.gov/. Accessed 24 Jul 2020.
Gilead Sciences. Gilead’s investigational antiviral remdesivir receives US Food and Drug Administration emergency use authorization for the treatment of COVID-19 [media release]. 1 May 2020. http://www.gilead.com.
Gilead Sciences. Veklury for intravenous injection 100mg: Japanese prescribing information. 2020. https://www.pmda.go.jp/. Accessed 24 Jul 2020.
Pharmaceuticals and Medical Devices Agency. Special approval for emergency on remdesivir for COVID-19 [media release]. 8 May 2020. https://www.pmda.go.jp/.
Food and Drug Administration. Taiwan Food and Drug Administration approves remdesivir to treat patients with severe COVID-19 disease [media release]. 11 Jun 2020. http://www.fda.gov.tw.
Gilead Sciences. European Commission grants conditional marketing authorization for Gilead’s Veklury® (remdesivir) for the treatment of COVID-19 [media release]. 3 Jul 2020. http://www.gilead.com/.
Health Sciences Authority. HSA grants conditional approval of remdesivir for treatment of COVID-19 infection [media release]. 10 Jun 2020. http://www.hsa.gov.sg.
Therapeutic Goods Administration. Australia’s first COVID treatment approved [media release]. 10 Jul 2020. http://www.tga.gov.au/.
The Korean Bizwire. S. Korea allows marketing authorization of remdesivir [media release]. 24 Jul 2020. http://www.firstwordpharma.com/.
Gilead Sciences Canada Inc. Health Canada grants marketing authorization with conditions (NOC/c) for Gilead’s Veklury® (remdesivir) for the treatment of coronavirus disease 2019 (COVID-19) [media release]. 28 Jul 2020. http://www.newswire.ca/.
Gilead Sciences Canada Inc. Product monograph: remdesivir for injection; remdesivir solution for injection. 2020. http://www.gilead.ca/. Accessed 30 Jul 2020.
Gilead Sciences Ireland UC. Veklury (remdesivir): EU summary of product characteristics. 2020. http://www.ema.europa.eu/. Accessed 24 Jul 2020.
The Arab Hospital Magazine. Gilead Sciences announces approval of Veklury® (remdesivir) in the United Arab Emirates for patients with severe COVID-19. 2020. http://thearabhospital.com/. Accessed 16 Jul 2020.
Jubilant Pharma. Jubilant Pharma Limited announces approval of ‘JUBI-R’ (remdesivir) in India for the treatment of COVID-19 [media release]. 20 Jul 2020. http://www.jubilantpharma.com/.
Somani VG. Approval of favipiravir tablets to Glenmark Pharmaceuticals and remdesivir injection to Cipla Ltd and Hetero Drugs. 2020. http://cdsco.gov.in/. Accessed 24 Jul 2020.
Mylan. Mylan secures regulatory approval for remdesivir lyophilized powder for injection 100 mg/vial in India for restricted emergency use in COVID-19 patients [media release]. 6 Jul 2020. http://newsroom.mylan.com/.
Gilead Sciences. Gilead submits New Drug Application to U.S. Food and Drug Administration for Veklury® (remdesivir) for the treatment of COVID-19 [media release]. 10 Aug 2020. http://www.gilead.com/.
Gilead Sciences. Company statements: Gilead Sciences statement on the initiation of clinical testing of an inhaled solution of remdesivir for potential outpatient treatment of COVID-19 [media release]. 8 Jul 2020. http://www.gilead.com/.
United States Securities and Exchange Commission. Form 8K: Ligand Pharmaceuticals Inc. 2015. http://sec.report/. Accessed 24 Jul 2020.
United States Securities and Exchange Commission. Form 10K: Ligand Pharmaceuticals Inc. 2016. http://investor.ligand.com/. Accessed 24 Jul 2020.
Ligand Pharmaceuticals. Ligand provides a corporate update and announces May 6th as the date for first quarter earnings call [media release]. 6 Apr 2020. http://www.ligand.com.
Gilead Sciences. Voluntary licensing agreements for remdesivir. 2020. http://www.gilead.com/. Accessed 24 Jul 2020.
Gordon CJ, Tchesnokov EP, Feng JY, et al. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J Biol Chem. 2020;295(15):4773–9.
Gordon CJ, Tchesnokov EP, Woolner E, et al. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J Biol Chem. 2020;295(20):6785–97.
Saha A, Sharma AR, Bhattacharya M, et al. Probable molecular mechanism of remdesivir for the treatment of COVID-19: need to know more. Arch Med Res. 2020. https://doi.org/10.1016/j.arcmed.2020.05.001.
Tchesnokov EP, Feng JY, Porter DP, et al. Mechanism of inhibition of Ebola virus RNA-dependent RNA polymerase by remdesivir. Viruses. 2019;11:4.
Pruijssers AJ, George AS, Schäfer A, et al. Remdesivir potently inhibits SARS-CoV-2 in human lung cells and chimeric SARS-CoV expressing the SARS-CoV-2 RNA polymerase in mice. bioRxiv. 2020. https://doi.org/10.1101/2020.04.27.064279.
Williamson BN, Feldmann F, Schwarz B, et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature. 2020. https://doi.org/10.1038/s41586-020-2423-5.
Mansuri Z, Shah B, Zafar MK, et al. Remdesivir and potential interactions with psychotropic medications: a COVID-19 perspective. Prim Care Companion CNS Disord. 2020;22(3):20com02664.
Humeniuk R, Mathias A, Cao H, et al. Safety, tolerability, and pharmacokinetics of remdesivir, an antiviral for treatment of COVID-19, in healthy subjects. Clin Transl Sci. 2020. https://doi.org/10.1111/cts.12840.
Yang K. What do we know about remdesivir drug interactions? Clin Transl Sci. 2020. https://doi.org/10.1111/cts.12815.
Gilead Sciences. Gilead announces results from phase 3 trial of remdesivir in patients with moderate COVID-19 [media release]. 1 Jun 2020. http://www.gilead.com.
Goldman JD, Lye DCB, Hui DS, et al. Remdesivir for 5 or 10 days in patients with severe Covid-19. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2015301.
Olender SA, Perez KK, Go AS, et al. Remdesivir for severe COVID-19 versus a cohort receiving standard of care [abstract no. 3960 and poster]. In: AIDS 2020: COVID-19 conference.
Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid-19—preliminary report. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2007764.
Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395(10236):1569–78.
Maserati R. Exposure to remdesivir through compassionate use: safety and efficacy in 163 patients [abstract no. 3948]. In: AIDS 2020: COVID-19 conference.
Grein J, Ohmagari N, Shin D, et al. Compassionate use of remdesivir for patients with severe Covid-19. N Engl J Med. 2020;382(24):2327–36.
Burwick R, Yawetz S, Stephenson KE, et al. Compassionate use of remdesivir in pregnant women with severe COVID-19 [abstract no. 3944 and poster]. In: AIDS 2020: COVID-19 conference.
Chiotos K, D. TP, Goldman DL, et al. Compassionate use of remdesivir in children with severe COVID-19 [abstract no. 3946 and poster]. In: AIDS 2020: COVID-19 conference.
Acknowledgements
During the peer review process, the manufacturer of remdesivir was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and conflicts of interest
Yvette Lamb is a salaried employee of Adis International Ltd/Springer Nature and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate and consent for publication, Availability of data and material, Code availability
Not applicable.
Additional information
Enhanced material for or this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.12752432/.
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Rights and permissions
About this article

Cite this article
Lamb, Y.N. Remdesivir: First Approval. Drugs 80, 1355–1363 (2020). https://doi.org/10.1007/s40265-020-01378-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01378-w