
Abstract
Epigenetics refers to the heritable regulation of gene expression through modification of chromosomal components without an alteration in the nucleotide sequence of the genome. Such modifications include methylation of genomic DNA as well as acetylation, methylation, phosphorylation, ubiquitination, and SUMOylation of core histone proteins. Recent genetic and biochemical analyses indicate that epigenetic changes play an important role in the development of cardiac hypertrophy and heart failure, with dysregulation in histone acetylation status, in particular, shown to be directly linked to an impaired contraction ability of cardiac myocytes. Although such epigenetic changes should eventually lead to alterations in the expression of genes associated with the affected histones, little information is yet available on the genes responsible for the development of heart failure. Current efforts of our and other groups have focused on deciphering the network of genes which are under abnormal epigenetic regulation in failed hearts. To this end, coupling chromatin immunoprecipitation to high-throughput profiling systems is being applied to cardiac myocytes in normal as well as affected hearts. The results of these studies should not only improve our understanding of the molecular basis for cardiac hypertrophy/heart failure but also provide essential information that will facilitate the development of new epigenetics-based therapies.
Similar content being viewed by others

What is “epigenetics”?
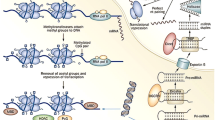
The eukaryotic genome is tightly compacted as a result of its association with highly conserved histone proteins. The interaction of stretches of genomic DNA with core histone proteins (two molecules each of H2A, H2B, H3, and H4) thus results in the formation of nucleosomes, which are the basic structural units of chromatin. The further association of histone H1 and other proteins with nucleosomes strengthens the compaction and gives rise to the highly ordered, condensed structure of the chromosome (Fig. 1). The interaction of genomic DNA with these chromosomal proteins greatly influences the access of transcriptional factors to their target DNA sequences and thereby regulates transcriptional activity.

Epigenetic changes at different levels of chromatin structure. CpG sites within genomic DNA undergo methylation, and core histones in nucleosomes undergo acetylation (Ac), methylation (Met), ubiquitination (Ub), sumoylation (S), or phosphorylation (P). Higher order chromatin structure is also dynamically modified by chromatin-remodeling complexes
“Epigenetics” refers to the heritable regulation of gene expression through the modification of chromosomal components without an alteration in the nucleotide sequence of the genome [1]. Several such modifications have been linked to the regulation of gene expression, including methylation of genomic DNA as well as acetylation, methylation, phosphorylation, ubiquitination, and sumoylation of histone proteins (Fig. 1).
Core histones have an amino-terminal tail that protrudes from the chromatin fiber and which is believed to interact with DNA or other histone or modulatory proteins. Lysine and arginine residues within this tail are the main targets for histone modification. Lysine-9 in histone H3 (H3-K9), for example, becomes methylated or acetylated in response to a variety of signals. In general, the acetylation of histones is associated with the induction of gene expression (Fig. 2). The acetylation of histone tails is thought to neutralize the positive charge of lysine residues and thereby to induce a decondensation of chromatin structure. The resulting open architecture of the chromosome allows transcriptional factors to access their binding sites in genomic DNA and to activate transcription. However, the same histone modification has been associated with seemingly diverse outputs in a context-dependent manner. The existence of a “histone code” has thus been proposed, with a combination of histone modifications – and not only one – supposedly specifying the outcome in terms of gene expression [2]. However, this hypothesis has been challenged by recent data [3].

Epigenetic changes and transcriptional activity. Suppression of gene expression (OFF) is correlated with the methylation (Met) of genomic DNA, deacetylation of histones, and methylation of H3-K9. In contrast, activation of gene expression (ON) is associated with unmethylated genomic DNA, acetylated (Ac) histones, and methylated H3-K4
The acetylation of histone tails is mediated by histone acetyltransferases (HATs), whose activity in cells is rapidly counteracted by that of histone deacetylases (HDACs) [4]. The turnover time of histone acetylation in cells is thus as short as a few minutes [5]. The importance of histone acetylation in the regulation of gene expression has been demonstrated for a variety of cellular processes, including cell differentiation, cell cycle progression, DNA repair, and carcinogenesis [6, 7].
Epigenetic status in cardiac myocytes
The regulation of histone acetylation has been shown to be linked to cardiac hypertrophy. The HAT activity of CREB-binding protein (CBP) and p300 is thus required for the induction of hypertrophic changes in cardiac muscle cells by phenylephrine [8]. Consistent with this observation, the inhibition of HDAC activity results in an increase in the size of muscle cells [9]. Furthermore, class II HDACs (HDAC4, -5, -7, and -9) suppress cardiac hypertrophy, in part by binding to and inhibiting the activity of myocyte enhancer factor 2 (MEF2) [10]. In contrast, however, HDAC2 together with Hop, a homeodomain protein, was found to promote cardiac hypertrophy in vivo in a manner sensitive to systemic administration of the HDAC inhibitor trichostatin A (TSA) [11]. Moreover, HDAC inhibitors prevent hypertrophy and sarcomere organization in cultured cardiac myocytes [12], which is suggestive of a positive role for HDACs in cardiac hypertrophy.
These seemingly discrepant findings may be attributable either to differential actions of different classes of HDACs (and, possibly, of HATs) with regard to myocyte hypertrophy or to a dissociation between the deacetylase activity of HDACs and a pro-hypertrophic function [10]. Clarification of the role of HATs and HDACs in hypertrophy would be facilitated by the identification of the genes targeted by these enzymes during the induction of hypertrophic changes. 1 Little is known, however, of the genes regulated by HATs or HDACs in myocytes. Induction of the atrial natriuretic peptide (ANP) gene is associated with the acetylation of histones (H3 and H4) located in the 3′ untranslated region of the gene [13]. Histones bound to the β-myosin heavy chain gene have also been shown to be targeted by HATs in myocytes [10].
Differential chromatin scanning (DCS) method
Given the essential role of histone acetylation in cardiac hypertrophy, it is important that the genes or genome regions bound to histones with such differential modifications be identified. Chromatin immunoprecipitation (ChIP) coupled with hybridization to genome tiling microarrays (“ChIP-on-chip” experiments) has been used to screen for those genes under epigenetic regulation [14–16]. However, an extensive mapping of ChIP products on the human genome has been hampered by insufficient information on human genome annotation. Furthermore, hybridization of genome-derived fragments to microarrays is prone to non-specific signals that partly represent the GC contents of the fragments.
To effectively isolate genome fragments with differential epigenetic regulation, we have developed a novel “DCS” method which basically couples ChIP to subtraction PCR [17, 18]. The DCS procedure is schematically shown in Fig. 3. Following the cross-linking of DNA to histones through the use of formaldehyde, both tester and driver cells are separately lysed and subjected to mild DNA shearing by sonication for a short period. Complexes of DNA and acetylated histones are then specifically immunoprecipitated with antibodies to acetylated histone H3, after which the DNA fragments are released from such complexes into solution.

The differential chromatin scanning (DCS) method. DNA fragments bound to acetylated (Ac) histones are purified by immunoprecipitation (IP) and subjected to TAG adaptor ligation (green bars) and PCR amplification. The tester DNA is then digested with XmaI, ligated to the first subtraction adaptor (red bars), and annealed with an excess amount of the driver DNA. Given that only the tester-specific fragments self-anneal, PCR with the first subtraction primer selectively amplifies these fragments. The products are subjected to a second round of subtraction PCR with the second subtraction adaptor and primer to ensure the fidelity of the subtraction. Reproduced from Kaneda et al. [17]
The nonspecific binding of residual RNA is then minimized by treating the DNA solution with RNase A, and the DNA fragments are then blunt-ended. The DNA is digested maximally with RsaI to obtain fragments with a relatively uniform size of several hundred base pairs. A TAG adaptor is ligated to both ends of the DNA fragments, and subsequent PCR amplification with a TAG primer yields amplicons with an XmaI/SmaI site at each end.
The tester DNA is then digested with XmaI (thereby generating cohesive ends), whereas the driver DNA is digested with SmaI (generating blunt ends). The tester DNA is ligated with the first subtraction adaptor [19] at its cohesive ends and is then annealed to an excess amount of the driver DNA. Under this condition, DNA fragments present only in the tester sample undergo self-annealing and thereby generate a binding site for the first subtraction primer at both ends. Subsequent PCR amplification with this primer thus selectively amplify the tester-specific DNA fragments [17].
To exclude DNA fragments that possess endogenous (probably nonspecific) binding sites for the first subtraction primer, we then digest the first subtraction products with XmaI and ligate the resulting molecules with the second subtraction adaptor. A second round of subtraction amplification is then performed with the second subtraction primer, yielding DNA fragments that are associated with acetylated histones, specifically in the tester cells [17].
To verify the fidelity of DCS, we attempted to isolate genome fragments which are the targets of HDAC in cardiac myocytes. A rat cardiomyocyte cell line, H9C2, was treated with TSA and was used as the tester, while the cells without the TSA treatment was used as the driver. Differential chromatin scanning was applied to this pair of cells and subsequently identified hundreds of genome fragments that could be immunoprecipitated by antibodies to acetylated histones only in the tester cells.
Some of the clones thus identified correspond to loci within or close to rat genes whose products function in intracellular calcium mobilization or antioxidant processes. One such clone, H9C2T-2_D09, mapped to a region encompassing intron 21 and exon 22 of Itpr3 (Fig. 4a), which encodes a receptor for inositol 1,4,5-trisphosphate that plays an important role in Ca2+-mediated signal transduction [18]. The cytosolic concentration of Ca2+ directly regulates muscle contraction and cardiac rhythm and is a determinant of myocyte hypertrophy and heart failure [20]. The amount of the genomic fragment corresponding to the H9C2T-2_D09 clone was 6.6-fold greater in the ChIP product of TSA-treated cells than in that of untreated cells (Fig. 4b), indicating that the extent of histone acetylation in this region of the genome of the tester cells was 6.6-fold that in the driver cells. Furthermore, inhibition of HDAC activity was accompanied by an increase in the amount of Itpr3 mRNA [18] (Fig. 4c). These data suggest that HDAC actively deacetylates a chromosomal region corresponding to Itpr3 and thereby suppresses the transcriptional activity of the gene.

Identification of Itpr3 as a target of histone deacetylase (HDAC) in cardiomyocytes. a One of the DCS clones (H9C2T-2_D09; red rectangle) was mapped to chromosome 20p12, spanning intron 21 and exon 22 of Itpr3. Exons are denoted by black boxes, the arrow indicates the direction of transcription, and blue triangles depict distance markers separated by 50 kbp. b Chromatin immunoprecipitates were prepared from H9C2 cells treated (+) or not (−) with 300 nM inhibitor trichostatin A (TSA) for 24 h. The amount of DNA corresponding to the H9C2T-2_D09 sequence in each chromatin immunoprecipitation (ChIP) product relative to that in the corresponding original sample before immunoprecipitation (PreIP) was then determined by real-time PCR. c The amount of Itpr3 mRNA relative to that of Gapdh mRNA in H9C2 cells treated or not with TSA was determined by quantitative reverse transcription (RT)-PCR. All data are means ± SD of triplicates from representative experiments that were performed at least twice. P values for the indicated comparisons were determined by Student’s t test. Reproduced from Kaneda et al. [18]
To visualize directly the genome-wide distribution of HDAC targets, we mapped the DCS genomic clones whose chromosomal positions were known to rat chromosome figures (Fig. 5). The HDAC targets were distributed widely throughout the rat genome, although some “hot spots” for deacetylation were apparent. For example, seven of the DCS clones mapped to chromosomal position 5q36, and detailed mapping revealed that all of these clones were located within a region spanning 27 Mbp. It is thus possible that regional alterations of chromatin structure result in coordinated transcriptional regulation of genes within the affected region.

Chromosomal distribution of HDAC targets. The genome fragments (red dots) isolated by the DCS method were mapped to rat chromosomes. Reproduced from Kaneda et al. [18]
Future directions
As described herein, cells manifest numerous types of epigenetic modifications, including acetylation and methylation, on core histone proteins. Although the results of biochemical and genetic studies suggest that histone acetylation plays an important role in the development of cardiac hypertrophy/heart failure (at least, in mouse), it is an open question whether changes in the other epigenetic marks are essential or, rather, causative of the heart disorders. An analysis of human heart specimens would be of particular great value.
In terms of technology development, DCS is not free from disadvantages. Although DCS can isolate genome fragments that are the recipients differential regulation of any epigenetic marks (provided specific antibodies are available), it does not measure the extent of “differential regulation”. In other words, DCS is more a qualitative approach than a quantitative one. Several high-throughput sequencing technologies are currently emerging which simultaneously provide sequence information for millions of clones [21]. Coupling such sequencing system to ChIP would be one of the ideal ways to quantitatively measure epigenetic modifications: (1) frequency in the read data would be a surrogate marker for the intensity of epigenetic modifications; (2) sequence information of the reads would be useful to map such reads onto human genome.
It is apparent that epigenetic change is the key event in the development of cardiac hypertrophy/heart failure. Analysis of human specimens with emerging technologies would substantially facilitate researchers in their efforts to pinpoint the causative genes for these disorders.
References
Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–63.
Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5.
Dion MF, Altschuler SJ, Wu LF, Rando OJ. Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci USA. 2005;102:5501–6.
Verdin E, Dequiedt F, Kasler HG. Class II histone deacetylases: versatile regulators. Trends Genet. 2003;19:286–93.
Waterborg JH. Dynamics of histone acetylation in vivo. A function for acetylation turnover? Biochem Cell Biol. 2002;80:363–78.
Kouzarides T. Histone acetylases and deacetylases in cell proliferation. Curr Opin Genet Dev. 1999;9:40–8.
Yasui W, Oue N, Ono S, Mitani Y, Ito R, Nakayama H. Histone acetylation and gastrointestinal carcinogenesis. Ann NY Acad Sci. 2003;983:220–31.
Gusterson RJ, Jazrawi E, Adcock IM, Latchman DS. The transcriptional co-activators CREB-binding protein (CBP) and p300 play a critical role in cardiac hypertrophy that is dependent on their histone acetyltransferase activity. J Biol Chem. 2003;278:6838–47.
Iezzi S, Di Padova M, Serra C, Caretti G, Simone C, Maklan E, et al. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev Cell. 2004;6:673–84.
Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–88.
Kook H, Lepore JJ, Gitler AD, Lu MM, Wing-Man Yung W, Mackay J, et al. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J Clin Invest. 2003;112:863–71.
Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, et al. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem. 2003;278:28930–7.
Kuwahara K, Saito Y, Ogawa E, Takahashi N, Nakagawa Y, Naruse Y, et al. The neuron-restrictive silencer element-neuron-restrictive silencer factor system regulates basal and endothelin 1-inducible atrial natriuretic peptide gene expression in ventricular myocytes. Mol Cell Biol. 2001;21:2085–97.
Weinmann AS, Yan PS, Oberley MJ, Huang TH, Farnham PJ. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev. 2002;16:235–44.
Ballestar E, Paz MF, Valle L, Wei S, Fraga MF, Espada J, et al. Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. EMBO J. 2003;22:6335–45.
Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–81.
Kaneda R, Toyota M, Yamashita Y, Koinuma K, Choi YL, Ota J, et al. High-throughput screening of genome fragments bound to differentially acetylated histones. Genes Cells. 2004;9:1167–74.
Kaneda R, Ueno S, Yamashita Y, Choi YL, Koinuma K, Takada S, et al. Genome-wide screening for target regions of histone deacetylases in cardiomyocytes. Circ Res. 2005;97:210–8.
Toyota M, Ho C, Ahuja N, Jair KW, Li Q, Ohe-Toyota M, et al. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res. 1999;59:2307–12.
Marks AR. Cardiac intracellular calcium release channels: role in heart failure. Circ Res. 2000;87:8–11.
Service RF. Gene sequencing. The race for the $1000 genome. Science. 2006;311:1544–6.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mano, H. Epigenetic abnormalities in cardiac hypertrophy and heart failure. Environ Health Prev Med 13, 25–29 (2008). https://doi.org/10.1007/s12199-007-0007-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12199-007-0007-8