
Abstract
The mechanisms regulating cell survival and thus its corollary, cell death, have been intensively studied over the last two decades. Recent studies have shed new light into how non-degradative ubiquitination of the kinase RIPK1 is critical in determining this cell fate. In this review, we summarize recent findings on how ubiquitination of RIPK1 constitutes a survival signal through both NFκB-independent and NFκB-dependent mechanisms. However, in the absence of ubiquitination, RIPK1 becomes a death-signaling molecule capable of engaging both the caspase-dependent apoptosis machinery and the recently described RIPK3-dependent necroptosis machinery. Another layer of complexity is now emerging in that components of the ubiquitin-modifying machinery are themselves regulated by proteolytic processing. This survival/death regulatory mechanism has been best analyzed in the context of TNF receptor signaling, but it is likely that principles learned from TNFR may be applicable to other immune receptors including the antigen and Toll-like receptors.
Similar content being viewed by others

Introduction
Innate and adaptive immunity is controlled by a multitude of different receptors that recognize ligands as diverse as antigenic peptides, cytokines and microbial products. The activation of different signaling complexes and transcription factors inside the cell determines its subsequent mode of action. Upon ligation of three main classes of immune receptors; the antigen receptors (AgR); Toll-like receptors (TLRs); and the tumor necrosis factor (TNF) death receptor superfamily, cell fate outcomes can be simplified to two choices: cell survival (which is essential for all subsequent differentiation and functions) or cell death. For example, during lymphocyte development, the vast majority of T cells die in the thymus as a result of negative selection, which removes cells expressing TCRs that react with self-tissue. A similar decision between cell survival and cell death occurs in mature T cells activated by specific antigen during the processes of T-cell expansion and contraction, which are regulated by the TCR and members of the TNF death receptor superfamily. In this review, we will discuss how NFκB and ubiquitination operate as two major pro-survival signaling mechanisms that control cell fate in the immune system, with particular emphasis on our work examining the regulation of receptor interacting protein kinase 1 (RIPK1)-mediated cell death programs in the TNF receptor pathway.
IKK complex and NFκB activation
Five transcription factors comprise the NFκB family, which regulates the expression of a huge variety of immune-related genes, such as inflammatory mediators, adhesion molecules, cytokines and major histocompatibility complex (MHC) [1]. Immunologists are thus very familiar with the role of NFκB in driving differentiation programs downstream of immune receptors. For example, the initial phase of TCR ligation triggers activation of NFκB, which acts in conjunction with other transcription factors to induce IL-2 synthesis and drive the expansion of T cells [2]. NFκB also promotes cell survival and thus keeps cells alive while they are performing particular effector functions during an immune response [3]. So how is NFκB activated? NFκB transcription factors are held in a state of suspended animation in the cytoplasm by their constitutive interaction with inhibitors of NFκB (IκBs): IκBα is the founding and the most well-understood member of this group of inhibitors. NFκB dimers are bound tightly to IκBs in the cytoplasm, which masks a nuclear localization signal within the NFκB proteins and thus prevents the NFκB transcription factors from entering the nucleus [4]. In order for NFκBs to enter the nucleus, the IκB proteins have to be removed. Removal of the IκBs occurs when immune receptors activate the IκB kinase (IKK) complex, a multisubunit complex comprised of IKKα, IKKβ and IKKγ (which we will refer to by its alternative name NFκB essential modifier or NEMO) [5]. Activated IKK complexes phosphorylate IκBα on serines 32 and 36, which leads to its subsequent ubiquitination and degradation by the proteasome. Degradation of IκBα unmasks the nuclear localization signal in the NFκB proteins, and they are now able to translocate to the nucleus and initiate specific gene expression programs. The downstream effects of NFκB activation are determined by the particular subsets of NFκB dimers activated [6, 7], in conjunction with the activation of other transcription factors. Therefore, the type of immune receptor activated and the cell type have a role to play in determining the outcome of IKK activation.
IκBα ubiquitination and NFκB activation
The degradation of IκBα is one of the foremost examples of how the post-translational modification called ubiquitination controls signaling events. Ubiquitin is a small 7-kDa protein that can be covalently attached to other proteins through an increasingly complex variety of conformations [8]. The first step in a ubiquitination reaction is the priming of ubiquitin molecules by the E1 enzyme (of which mammalian cells have two isoforms). The C-terminal glycine of ubiquitin forms a highly unstable thioester bond with a cysteine in the active site of the E1 enzyme. The ubiquitin molecules can then be shuttled to a cysteine in the active site of an E2 enzyme. In the case of IκBα, the E2 enzymes include members of the Ubc family [9]. The third step is the transfer of the ubiquitin molecules to a substrate protein, which is orchestrated by a third class of enzymes or E3 ligases. In the case of IκBα ubiquitination, the Fbox protein βTRCP functions as the E3 enzyme and forms a reactive thioester intermediate with the ubiquitin molecule after its transfer from the Ubc E2 enzymes [10]. βTRCP forms a complex with the proteins Skp1 and Cul1, and this complex specifically recognizes the phosphorylated form of IκBα, therefore providing a critical link between the activation of the IKK complex and the ubiquitination of IκBα [4]. The E3 enzyme catalyzes the formation of a peptide bond between the glycine of the ubiquitin molecule and the amino side chain of a lysine residue in the target protein. E3 enzymes can then catalyze the attachment of subsequent ubiquitin molecules to the initial ubiquitin molecule covalently linked to the acceptor lysine of the substrate protein, leading to the formation of polyubiquitin chains. Ubiquitin has seven lysine residues itself, all of which are capable of forming a peptide bond with another ubiquitin molecule and seeding a polyubiquitin chain. In the case of IκBα, the acceptor lysines 21 and 22 in IκBα are conjugated to ubiquitin, and then, a polyubiquitin is formed with all subsequent ubiquitin molecules conjugated via lysine 48 of the preceding ubiquitin molecule. K48 polyubiquitination is an extremely important post-translational modification that regulates signaling processes, as it targets proteins to the proteasome for degradation. In the case of IκBα, this degradation is a key step in the activation of NFκB by removing the inhibitor that sequesters NFκB in the cytoplasm. Expression of an IκBα protein in which the phosphorylation sites are mutated to alanine is sufficient to prevent phosphorylation and degradation of endogenous IκBα [11, 12]. Therefore, IκBαS32A,S36A functions as a super-repressor, blocking all NFκB-mediated gene transcription.
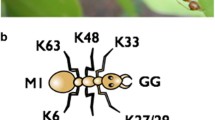
In recent years, the complexity with which the ubiquitination reactions can occur is becoming clear, and there are examples of signaling proteins that are attached to one ubiquitin molecule (monoubiquitination), multiple ubiquitins with no chain formation (multi-monoubiquitination), polyubiquitin chains formed via any of the seven lysine residues and branched forms of mixed chain linkages [13]. Moreover, polyubiquitin chains can be formed by ubiquitin molecules conjugated from head to tail (linear ubiquitination). For example, the adaptor protein NEMO undergoes linear ubiquitination, and this maybe a key step in the activation of the IKK complex [14, 15]. As a general rule, K48-linked polyubiquitination serves to target proteins to the proteasome for degradation, whereas the other forms of ubiquitination regulate the activity of signaling molecules in a non-degradative manner. In this review, we will discuss how NFκB and ubiquitination control the formation of death-signaling complexes containing the kinase RIPK1 downstream of multiple receptors in the immune system but with particular emphasis on the TNF death receptor superfamily.
RIPK1 ubiquitination and NFκB activation
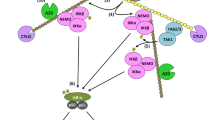
Firstly, to introduce how ubiquitination regulates the function of RIPK1, we will discuss the role of RIPK1 in the activation of NFκB by TNF receptor 1 (TNFR1). Ligation of TNFR1 by trimeric TNF leads to the recruitment of cytoplasmic adaptor molecules such as TRADD, TRAF2 and RIPK1. One of the earliest descriptions of this signaling complex included the observation that RIPK1 is heavily modified when recruited to TNFR1 [16], which suggested the possibility that multiple ubiquitin proteins may be attached to RIPK1. This modification occurred coincident with the recruitment and activation of the IKK complex and degradation of IκBα, suggesting that this modification might be important for controlling IKK activation. Indeed, Jurkat T cells rendered deficient in RIPK1 by chemical mutagenesis were unable to activate NFκB [17]. The observation that RIPK1 is essential for the activation of NFκB by TNFR1 was also seen in cells derived from RIPK1 knockout mice [18]. This modification of RIPK1 was shown to be polyubiquitination and required the presence of the RING E3 enzyme TRAF2 [19]. Activation of NFκB is only slightly compromised in TRAF2 knockout cells [20], which may call into question the absolute requirement for RIPK1 ubiquitination in the optimal activation of NFκB. However, loss of TRAF2 leads to heightened susceptibility to TNF-induced cell death [20]. The first study that provided excellent correlative evidence that ubiquitination of RIPK1 is important for the activation of IKK by TNFR1 is the finding that the enzyme A20 could inhibit activation of NFκB by ubiquitin-editing RIPK1 [21]. A20 was found to first remove K63-linked polyubiquitin chains from RIPK1 and then subsequently attach K48-linked polyubiquitin chains that target RIPK1 for degradation by the proteasome. In A20 knockout MEFs, RIPK1 becomes hyper-ubiquitinated and stays at the receptor with concomitant prolonged NFκB activity in the cells. Therefore, there was a clear correlation between non-degradative ubiquitination of RIPK1 and persistent activity of the IKK complex. In contrast, stable expression of A20 in human cells leads to reduced levels of RIPK1 at the TNFR1 and inhibition of NFκB responses [22]. Therefore, there was mounting circumstantial evidence that linked ubiquitination of RIPK1 and the activation of NFκB by TNFR1.
Definitive evidence that the non-degradative ubiquitination of RIPK1 is central to activation of the IKK complex came from three studies [23–25], which identified the ubiquitin acceptor site in RIPK1 and discovered that the IKK complex scaffolding adaptor protein NEMO contains an ubiquitin recognition domain. The Chen and Lin laboratories identified lysine 377 of RIPK1 as the acceptor site for K63-linked non-degradative polyubiquitin chains and showed that RIPK1-deficient Jurkat T cells that express RIPK1-K377R are unable to mount an efficient NFκB response when stimulated with TNF [23, 24]. It was already known that the TAB 2 and TAB 3 adaptor proteins contain zinc fingers that specifically interact with ubiquitinated RIPK1 [26], and the Chen and Lin groups demonstrated that TAB 2/TAB 3 and the TAK1 kinase were not recruited to non-ubiquitinated RIPK1-K377R [23, 24]. In addition, the Chen and Ashwell groups identified a ubiquitin-binding motif in NEMO that specifically recognizes K63-linked polyubiquitin chains [24, 25]. Similar to TAB 2/TAB 3-mediated recruitment of TAK1 to ubiquitinated RIPK1, NEMO-mediated recruitment of IKKα and IKKβ was dependent upon the ability of NEMO to recognize ubiquitinated RIPK1. NEMO and the IKK complex do not bind the RIPK1-K377R mutant; conversely, ubiquitinated RIPK1-WT does not bind IKK when the ubiquitin-binding motif is mutated in NEMO. Therefore, the activation of IKK and subsequent NFκB-mediated transcriptional programs downstream of TNFR1 depends on the specific recruitment of multiple components to ubiquitinated RIPK1: TAB 2/TAB 3 binding to ubiquitinated RIP1 forms a bridge to the TAK1 kinase, whereas NEMO binding forms a bridge for IKKα and IKKβ recruitment. Activation of the IKK complex downstream of other immune receptors does not use RIPK1 to connect ligated receptors to IKK complexes; however, the formation of the IKK complexes depends upon similar ubiquitination of key scaffolding molecules. For example, the ubiquitination of the kinase IRAK1 downstream of the TLRs [27, 28] or Malt1 [29] and Bcl10 [30] in the TCR pathway mediates recruitment and activation of the IKK complex. The ability of the adaptor proteins NEMO and TAB 2/TAB 3 to recruit IKK and TAK1 is crucial to the activation of NFκB, and thus the production of IL-2 by T cells ligated through the TCR [31], but RIPK1 does not seem to contribute to activation of NFκB by AgRs or TLRs. However, RIPK1 has an important role to play in connecting these immune receptors to the formation of cell death complexes in particular circumstances.
Ubiquitin ligases for RIPK1
Recent studies of RIPK1 ubiquitination demonstrate the complexity with which ubiquitination reactions can occur and how little we understand about how E3 enzymes operate. In the case of RIPK1, ubiquitination requires the presence of the RING domain E3 enzymes TRAF2 and the cIAPs [19, 32–34]. RING domain E3 ligases are different from E3 ligases like the SCF E3 complex that ubiquitinates IκBα, because they do not form a reactive thioester intermediate with activated ubiquitin molecules. RING domain E3 enzymes catalyze the transfer of ubiquitin from the conjugated E2 but do not form a direct reaction intermediate with the ubiquitin molecule itself. Therefore, it is less clear how RING E3 enzymes help maneuver ubiquitin-conjugated E2 in order to achieve attachment of ubiquitin to a specific lysine residue in a target protein. Similarly, it is unclear how RING domain E3s controls the formation of different types of polyubiquitin chain. There was plenty of evidence that cIAPs could catalyze the attachment of K63-linked polyubiquitin chains to RIPK1 in in vitro ubiquitination reactions [25, 34, 35]. Cells from cIAP knockout mice also display defective ubiquitination of RIPK1 at TNFR1, which could be restored by complementing the cells with wild-type cIAPs but not with RING-mutant cIAPs [33]. Similarly, degradation of the cIAPs induced by pharmacological agents impairs the ubiquitination of RIPK1 [34]. Together, these studies implicate the cIAPS as the E3 enzymes responsible for the non-degradative ubiquitination of RIPK1. However, ubiquitination of RIPK1 is also clearly defective in MEFs from TRAF2 knockout mice [19, 32]. RIPK1 ubiquitination in TRAF2 knockout MEFs can be restored by expression of wild-type TRAF2 but not RING-mutant TRAF2 [33, 36]; thus, loss of either TRAF2 or cIAP RING E3 function results in impaired ubiquitination of RIPK1. TRAF2 and the cIAPs interact: It has been proposed that TRAF2 is required to bind and recruit cIAPs to the TNFR1 complex and that cIAPS are the bona fide E3 ligases for RIPK1 [36]. This conclusion was based on the observation that TRAF2 is unable to attach K63-linked polyubiquitin chains to RIPK1 in in vitro ubiquitination reactions but that the cIAPs can readily catalyze the attachment of a variety of polyubiquitin chains to RIPK1. However, a recent study has shown that TRAF2 requires the presence of the lipid cofactor sphingosine-1-phosphate (S1P) in order to function as an E3 ligase for RIPK1 [37]. Ubiquitination of RIPK1, recruitment of IKK and activation of NFκB are abrogated in cells that lack sphingosine-1-kinase, the enzyme that catalyzes the production of S1P. Therefore, it seems that both TRAF2 and the cIAPs play an important role in mediating ubiquitination of RIPK1 in response to ligation of TNFR1. In conclusion, optimal activation of NFκB by TNFR1 requires a complex interplay between the RING E3 enzymes TRAF2 and the cIAPs, which catalyze non-degradative ubiqutination of RIPK1 but the precise function of either E3 remains unclear.
NFκB-independent pro-survival pathways?
Activation of NFκB had been known for a long time to prevent TNFR1 from inducing apoptosis [38]. Before we discuss the cooperation between NFκB and RIPK1 ubiquitination in preventing cell death, we will first revisit the seminal paper from the Tschopp lab that identified the NFκB and death-signaling complexes and their location. Micheau and Tschopp [16] showed that the TRADD, RIPK1 and TRAF2 adaptors are rapidly recruited to TNFR1 at the plasma membrane and subsequently mediate recruitment of the IKK complex; they designated this Complex I and suggested that this signaling complex at the plasma membrane orchestrates the activation of NFκB responses. At later time-periods of stimulation with TNF, they demonstrated that a different complex forms in the cytoplasm containing the death-signaling molecules RIPK1, FADD, caspase 8 and caspase 10. They designated this slow-forming aggregation of pro-apoptotic molecules Complex II. By over-expression of the IκBα super-repressor (IκBαSR), they revealed that the death-signaling Complex II forms regardless of whether or not cells are able to activate NFκB but that the presence of key antiapoptotic proteins in Complex II is abrogated by blockade of NFκB. In particular, expression of the IκBαSR reduced the amount of cFLIP, A20 and cIAPs in Complex II, proteins that were already known to prevent the activation of caspases and apoptosis [39]. This study suggested that ligation of TNFR1 does not generally induce cell death because activation of NFκB by Complex I leads to transcription of anti-apoptotic factors that subsequently prevent the activation of apoptosis by Complex II. However, there were several problems with this model implicating NFκB as the major mediator of cell survival.
First, it takes a certain amount of time for the activation of NFκB to lead to the transcription, translation and recruitment of newly synthesized NFκB-dependent anti-apoptotic factors to Complex II. During this time-period, TNFR1 is ligated for several hours and Complex I contains two death-domain proteins, TRADD and RIPK1, that are capable of stimulating apoptosis. Therefore, there must be mechanisms to keep cell death in check at early time-points also. Second, this model does not account for the role of RIPK1 in controlling cell survival and cell death. It had been known that RIPK1 could cause cell death upon over-expression in certain cell types [40], but in vivo RIPK1 was required for cell survival by activating NFκB and RIPK1 appeared dispensable for TNF-induced apoptosis [18]. However, it was known that RIPK1 was critical for the induction of cell death by ligation of the inducible TNF receptor: TNFR2. TNFR2 is an inducible receptor for TNF and can be expressed by T cells after TCR ligation and in other cell types after recognition of microbial products [41]. Ligation of TNFR2 leads to enhanced cell death upon TNFR1 ligation [42]. The enhanced cell death requires RIPK1 and occurs despite the fact that ligation of TNFR2 is a very potent stimulus of NFκB. Intriguingly, ligation of TNFR2 switches the activation of NFκB to a RIPK1-independent mechanism and simultaneously switches the induction of cell death to RIPK1-dependent mechanisms [43]. These effects of TNFR2 ligation closely mimic the original observation of the phenotype of TRAF2 knockout cells, which proposed that the RING E3 ligase TRAF2 has NFκB-independent cell protective effects [20]. The original report of the phenotype of TRAF2 knockout MEFs suggested that TRAF2 was a minor requirement for the full activation of NFκB but that it was required to prevent cell death [20]. TRAF2 knockout MEFs were more sensitive to TNF-induced cell death than their wild-type counterparts despite nearly equivalent activation of NFκB. This heightened sensitivity to TNF-induced cell death was also observed when the TRAF2 knockout MEFs were treated with cycloheximide, a protein synthesis inhibitor that prevents production of all new NFκB-dependent anti-apoptotic proteins. The suggestion that TRAF2 blocks TNF from triggering apoptosis by an NFκB-independent mechanism was backed up by studies showing that blockade of TRAF2’s E3 ligase activity by over-expression of a RING domain deleted TRAF2, which functions as a dominant negative (TRAF2DN), could enhance TNF-mediated apoptosis in cells that were unable to activate NFκB due to the expression of the IκBαSR [44, 45]. Together, these studies suggested that TRAF2 has potent pro-survival activity that does not depend on NFκB-mediated gene transcription. This was a somewhat surprising finding since NFκB had already been identified as a crucial pro-survival signal activated by TNFR1 in order to keep cells alive, for example, by inducing expression of anti-apoptotic molecules such as cFLIP, TRAF2, cIAPs, A20 and Bcl-xl. [39, 46–52]. Natoli et al. [53] originally proposed that TRAF2 may prevent a pro-death molecule from functioning upon TNFR1 ligation before NFκB-mediated pro-survival pathways were initiated. Ligation of TNFR2 had been reported to trigger autoubiquitination of the cIAPs and TRAF2, leading to their proteasomal degradation [54, 55]. Therefore, there appeared to be a correlation between loss of the RING domain E3 s for RIPK1 ubiquitination and the induction of RIPK1-dependent cell death programs. We hypothesized that non-degradative ubiquitination of RIPK1 may prevent cell death not only via activating NFκB, but also by preventing RIPK1 from engaging the death-signaling apparatus before the protective effects of NFκB activation come into play.
RIPK1 ubiquitination and cell death
In order to test whether non-degradative ubiquitination of RIPK1 by TRAF2 or the cIAPS in the TNFR1 complex may prevent RIPK1 from functioning as a pro-death molecule, we examined cell death responses in RIPK1-deficient Jurkats T cells stably expressing RIPK1-WT, RIPK1-K377R or an irrelevant control protein [56]. At early time-points, we found that there is no difference in cell death between cells that lack RIPK1 and cells that express RIPK1-WT, suggesting that activation of NFκB is not sufficient to prevent apoptosis in the early hours after TNFR1 ligation. Over a period of 24-h stimulation with TNF, however, RIPK1-WT-expressing T cells undergo less apoptosis than cells that lack RIPK1, suggesting that NFκB-mediated gene transcription contributes to an anti-apoptotic effect over the long term. In order to confirm that the protective effect of RIPK1 expression was due to NFκB activation, we blocked all NFκB-mediated gene expression by over-expression of the IκBαSR. As expected, when NFκB activity was absent, there was no protective effect of RIPK1 re-expression in the RIPK1-deficient Jurkats. These data are consistent with the loss of NFκB responses in RIPK1-deficient cells and their sensitization to TNF-induced apoptosis, presumably due to the lack of inducible expression of anti-apoptotic proteins that limit caspase activation in Complex II. So what happens when NFκB is prevented by expressing a RIPK1 protein that cannot be ubiquitinated? We observed that Jurkat T cells that stably express the RIPK1-K377R mutant protein start to undergo apoptosis at very early time-points, and this accelerated rate of apoptosis continues for at least 24 h. After 5 h of treatment with TNF, there is more cell death in RIPK1-K377R cells (which activate low levels of NFκB) than in RIPK1-null Jurkat T cells (which do not activate NFκB), indicating that there is little overall correlation between NFκB and protection from cell death. More convincingly, cells that express RIPK1-K377R undergo more apoptosis than RIPK1-WT-expressing cells when all NFκB responses are blocked by co-expression of the IκBαSR, which indicates that in the absence of non-degradative ubiquitination, RIPK1 begins to function as a pro-death molecule. The protective effect of RIPK1 ubiquitination thus does not depend entirely on NFκB and phenocopies the mechanism by which the RING E3 ligase TRAF2 appears to prevent cell death. More recent studies have shown that the loss of the cIAPs, either due to genetic ablation or pharmacological manipulation [34, 57], also leads to abrogation of RIPK1 ubiquitination and the conversion of RIPK1 to a pro-death-signaling molecule. These data generated from studies using different molecular biology trick to prevent RIPK1 ubiquitination dovetail precisely with the physiological trigger for TNFR1 to initiate apoptosis: ligation of TNFR2. As discussed above, TNFR2 ligation triggers the loss of TRAF2 and cIAPs; loss of these E3 ligases correlates with the ability of TNFR2 to greatly augment apoptosis triggered through TNFR1 despite the fact that ligation of TNFR2 is a very potent stimulus of NFκB [42, 43]. We propose that there are two main checkpoints that prevent TNF-induced cell death that are controlled by RIPK1 ubiquitination. At early time-points, RIPK1 ubiquitination prevents RIPK1 from triggering cell death, but this protective effect of RIPK1 ubiquitination does not require NFκB. However, the activation of NFκB by ubiquitinated RIPK1 leads to long-term protection from apoptosis by preventing the activation of caspases in Complex II.
So how does this ubiquitination process prevent RIPK1 from initiating apoptosis? We have subsequently shown that the binding of NEMO to the ubiquitin chains on RIPK1 prevents RIPK1 from binding caspase 8 [58]. Cells that lack NEMO are extremely susceptible to TNF-mediated apoptosis, more so than cells rendered NFκB-deficient by stable expression of the IκBαSR. The enhanced apoptosis observed as a result of the loss of NEMO was entirely RIPK1 dependent and could be blocked by the expression of wild-type NEMO, but not by that of NEMO mutants that are unable to recognize polyubiquitinated RIPK1. We would predict that stable formation of Complex I around ubiquitinated RIPK1 is a key factor in preventing RIPK1 from functioning as a pro-apoptotic molecule. Therefore, it is likely that recruitment of TAB 2/TAB 3 and TAK1 to ubiquitinated RIP1 is likely to contribute to the NFκB-independent cell protection and potentially the ubiquitination of other Complex I components by TRAF2, cIAPS and other E3 enzymes. The fact that loss of NEMO is a particularly potent trigger of RIPK1-dependent apoptosis further suggests that ubiquitination of NEMO itself is likely to contribute to maintaining cell survival. NEMO and other ubiquitin-binding proteins are key components of the signaling complexes that activate IKK and NFκB downstream of other immune receptors such as the TCR and TLRs. Therefore, it seems likely that the interaction of these adaptors with ubiquitinated RIPK1 may prevent these receptors from engaging RIPK1-dependent cell death mechanisms. Although most of these receptors do not require RIPK1 to trigger NFκB activity, it is clear that RIPK1 can mediate AgR [59–61]- and TLR-induced cell death [62], which suggests that the death-promoting activity of RIPK1 is normally retarded. For example, RIPK1 is required to prevent cell death in B cells stimulated through the TLR4 [63] but mediates LPS-induced cell death [64].
RIP kinases and programmed necrosis
As far as the immune system is concerned, activation of apoptosis by RIPK1 is not the only interesting or physiologically relevant form of cell death. It has been known for a long time that TNF can trigger cell death by necrosis [65, 66], a form of cell death that is anticipated to be pro-inflammatory due to the release of genomic DNA and other DAMPs [67, 68]. Necrosis, also called necroptosis, is a form of programmed cell death that occurs when the caspase-dependent programs of apoptosis are blocked. In particular, it had been known for a long time that caspase 8 actually prevents cell death by suppressing programmed necrosis [69–71]. The ability to switch on an alternative form of cell death when caspases are blocked has clear advantages to the immune system when we encounter viruses that encode caspase inhibitors and other proteins that prevent apoptosis. The best characterized example is the poxvirus Vaccinia which encodes Spi2, an inhibitor of both caspases 1 and 8 [72, 73] and is thus able to prevent the caspase-dependent maturation of IL-1, a major-pro-inflammatory cytokines. Caspase 11 has recently been identified as a key caspase for IL-1 production during certain types on infection [74]; it remains to be seen whether Spi2 can inhibit caspase 11. Cells that express the cowpox homologue of Spi2A (CrmA) are unable to undergo apoptosis due to blockade of caspase 8 activity [75]. However, in certain cell types, this blockade of caspase 8 cause cells to switch on an alternative necrotic cell death pathway called programmed necrosis or necroptosis that is mediated by RIPK1 [69, 71]. In vivo, the induction of programmed necrosis by vaccinia requires TNFR2 in addition to TNFR1 [66]. TNFR2-deficient mice that are infected with vaccinia do not mount an inflammatory response and are unable to control viral replication. This suggests that necrotic cell death triggered by vaccinia virus infection in vivo is essential to the antiviral response by triggering cell death of virus-infected cells and that the necrotic cell death may initiate an inflammatory response. This circumvents the strategy of the virus to block inflammation and apoptosis mediated by caspase 1 and caspase 8, respectively. Necrotic cells can be specifically recognized by DNGR1 and other DAMP receptors [76, 77]; DNGR1 recognition is critical for cross-presentation of cell-associated antigen and the mounting of a CD8 T-cell response. In addition, targeting antigen to DNGR1 greatly augments antibody production [78]. It remains to be examined whether programmed necrosis of virus-infected cells contributes to the activation of adaptive immune responses to Vaccinia virus.
The signaling machinery that regulates necrotic cell death has been the focus of intense research over the past few years. The Chan, Wang and Zhang labs all pinpointed the kinase RIPK3 as a key mediator of programmed necrosis [79–81]. RIPK3 is related to RIPK1: It contains a similar kinase domain but lacks the death domain [82]. Indeed, RIPK3 knockout mice phenocopy the TNFR2 knockout mice with respect to Vaccinia virus infection [81], confirming that programmed necrosis is a key component of the antiviral immune response. However, it is becoming increasingly clear that aberrant programmed necrosis can be immunopathological and has to be repressed during various developmental processes. The studies from the Wang [79] and Zhang [80] groups provided the first indication that necrotic cell death mediated by RIP kinases has the potential to be inflammatory even in the absence of a microbial product. RIPK3 knockout mice were more resistant to cerulein-induced pancreatitis, an acute inflammatory condition drive by TNF. The ability of caspase 8 to prevent necrosis has recently been shown to be crucial for the survival of cells during embryogenesis and hematopoiesis. Caspase 8 knockout mice are not viable, but surprisingly, this lethality could be entirely rescued by crossing the mice onto a RIPK3 knockout background [62, 83]. Therefore, programmed necrosis has to be turned off by caspase 8 during embryonic development. Lymphocytes that lack caspase 8 and FADD undergo premature cell death upon ligation of their TCR [59, 60]. This cell death was not blocked by caspase inhibitors but could be blocked by necrostatins: inhibitors of RIPK1 kinase activity. Similarly, premature death of caspase 8- or FADD-deficient lymphocytes is prevented by genetic deletion of RIPK1 [84] or RIPK3 [62, 83], indicating that the ability of caspase 8 to suppress RIPK-mediated necrosis is key to the initial expansion phase of the T-cell response. This suggests that in the absence of caspase 8 activity, RIPK1 is engaged as a pro-death molecule by the AgRs. The accumulation of T cells in the absence of caspase 8 or FAS but not in RIPK3 knockout mice suggests that apoptosis and FAS may mediate the contraction phase of the T-cell response. The lymphocytes that accumulate in the caspase 8 or FAS knockouts have an unusual surface phenotype so it is hard to establish whether loss of apoptosis has contributed to increased survival of memory T cells or increased survival of effector cells that would normally die during the contraction phase. Therefore, RIPK-dependent necrotic cell death mechanisms are crucial regulators of lymphocytes in vivo.
The importance of programmed necrosis as an inflammatory cell death process has also been shown by the spontaneous development of inflammatory bowel disease in mice that lack caspase 8 or FADD in the gut epithelium [85, 86]. Moreover, inflamed tissue from Crohn’s disease patients reveals that necrotic intestinal epithelial cells can also be observed in chronic inflammatory conditions in humans. Similarly, cell-specific knockout of FADD in the skin leads to inflammation similar to the chronic condition psoriasis [87]. In all these examples, the inflammation corresponding to the presence of necrotic cells could be prevented by crossing the mice onto the RIPK3 knockout background. Intestinal inflammation could be abrogated by antibiotic treatment, suggesting that microbial products have a part to play in necrosis-driven inflammation, consistent with earlier studies that bacterial products such as LPS which ligates TLR4, or activation of the NOD receptors for intracellular PAMPs, can initiate necrotic cell death. However, FADD-deficient Paneth cell death and enteritis were not rescued by the elimination of the microbiota, indicating that some cell types may respond to an endogenous ligand in order to undergo necrosis. Despite the importance of caspase 8 in keeping cells alive during embryogenesis, hematopoiesis and T-cell activation, it was unknown what critical substrate caspase 8 cleaves in order to prevent necrotic cell death.
Inflammation in caspase 8 and FADD knockout mice could also be abrogated by removing another enzyme that is required for RIPK1 and RIPK3 to trigger programmed necrosis: the deubiquitinase enzyme CYLD. CYLD was originally shown in a genetic screen to be required for necrotic cell death of L929 cells [88], a mouse fibrosarcoma cell line that is particularly susceptible to necrosis upon treatment with caspase inhibitors and TNF. CYLD is a deubiquitinase that can remove K63-linked non-degradative polyubiquitin molecules from multiple targets, and so far, this has been shown to enable CYLD to block IKK-mediated signaling complexes, for example, activation of NFκB by TCR or TLRs [89, 90]. Surprisingly, CYLD had not been shown to function as a particularly good inhibitor of NFκB activated by TNFR1. Our recent data shed light on this phenomenon by identifying CYLD as the key substrate processed by caspase 8 in order to prevent necrosis downstream of TNFR1.
RIPK1 deubiquitination and cell death
As discussed above, ubiquitination of RIPK1 has a profound effect on its ability to mediate NFκB and cell death programs, which suggested that deubiquitinase enzymes that can remove ubiquitin chains from RIPK1 are likely to be the important modulators of death signaling by RIPK1. Two enzymes that are able to deubiquitinate RIPK1 have been identified thus far: A20 [21] and CYLD [91]. Both A20 and CYLD are capable of removing K63-linked polyubiquitin chains from RIPK1, but they have striking differences in their effects on cell death pathways. A20 is recruited to the TNFR1 complex in the plasma membrane and removes the K63-linked polyubiquitin chains that form the backbone for the formation of the IKK complex. As expected, MEFs from A20-deficient MEFs have hyper-ubiquitinated RIPK1 bound to TNFR1 and activate much more NFκB [21], which may contribute to the chronic inflammatory conditions seen in these mice [49]. However, A20 is also a potent inhibitor of TNF-induced apoptosis. At first, this may seem at odds with the data suggesting that blockade of RIPK1 ubiquitination leads to RIPK1 rapidly forming a complex with caspase 8 and enhancing apoptosis and the fact that A20 is a potent inhibitor of the anti-apoptotic NFκB program. However, A20 is a multitasking ubiquitin-editing enzyme. The removal of the K63-linked polyubiquitin chains on RIPK1 by the deubiquitinase domain of A20 occurs concurrently with the K48-linked polyubiquitination of RIPK1 by zinc fingers in A20 that function as an E3 enzyme. Therefore, it appears that A20 is able to block both NFκB and apoptosis by completely removing RIPK1 from the TNFR1 complex [21]. Apoptosis mediated by NFκB blockade does not require RIPK1: RIPK1-deficient Jurkats die by apoptosis when treated with TNF, and this is completely blocked by A20 (unpublished data), which suggests that A20 may target other pro-apoptotic enzymes in order to prevent apoptosis initiated by Complex II. Blockade of caspase 8 would normally trigger a switch to RIPK1-dependent necrotic cell death but over-expression of A20 completely prevents cell death in NEMO-deficient Jurkats [22], suggesting that degradation of RIPK1 by A20 is probably sufficient to keep necrotic cell death in check. Therefore, A20 functions as a pro-survival molecule by preventing RIPK1-independent apoptosis and by blocking all RIPK1-dependent apoptotic and necrotic cell death. A20 also can prevent necrosis in response to other TNF superfamily death ligands and complement [92]. In response to oxidative stress, however, the blockade of NFκB by A20 may sensitize the cells to necrotic cell death [93]. The phenotype of A20 knockout mice has been attributed to the inability to downregulate pro-inflammatory NFκB signaling by TNF and TLRs [49, 94], but it remains to be seen whether or not the inability to regulate pro-inflammatory necrotic cell death may also contribute to the chronic inflammation. Clearly, since A20-deficient mice are viable, A20 has no role to play in preventing necrosis during embryonic development. Caspase 8 and FADD appear to be the major players in suppressing necrosis during embryogenesis.
Caspase 8 removes CYLD deubiquitinase
So how do caspase 8 and FADD prevent necrosis? We have recently solved this mystery by identifying CYLD as the key substrate for caspase 8 to prevent RIPK-mediated programmed necrosis [95]. CYLD is a deubiquitinase that, like A20, is capable of removing K63-linked polyubiquitin chains from RIPK1 and many other targets. In the TCR and TLR signaling pathways, the removal of non-degradative ubiquitin chains by CYLD on key substrates such as NEMO appears to prevent activation of the IKK complex by multiple immune receptors. However, there is very little effect of CYLD deficiency on TNF-induced NFκB, suggesting that A20 is the primary deubiquitinase that regulates RIPK1’s NFκB signaling. CYLD is normally unable to function as a deubiquitinase toward RIPK1 in the TNFR1 pathway because it is rapidly cleaved by caspase 8. We find that ligation of TNFR1 leads to the rapid processing of CYLD at aspartate 215. A 25-kDa fragment corresponding to the N-terminal cleavage product is readily detectable by Western blot but the C-terminal cleavage product that would contain the deubiquitinase activity of CYLD is rapidly lost. The C-terminal portion of CYLD contains a putative PEST domain, and the processing of CYLD by caspase 8 is accompanied by the rapid proteasomal removal of the C-terminus cleavage product of CYLD containing the deubiquitinase domain. This indicates that CYLD is normally unable to function as a deubiquitinase for RIPK1 in the presence of caspase 8 activity. However, when caspase 8 activity is blocked by either genetic deletion or pharmacological inhibition, CYLD is stabilized and can remove polyubiquitin chains from RIPK1. CYLD knockout MEFs that express the non-cleavable mutant CYLD-D215A rapidly undergo RIPK1- and RIPK3-mediated necrotic cell death without the need for caspase 8 inhibition. In the CYLD-D215A-expressing cells, no processing of CYLD occurs, and RIPK1 is more rapidly deubiquitinated and disengages from NEMO. Therefore, when caspase 8 is unable to process CYLD and remove its deubiquitinase domain, RIPK1 is deubiquitinated by CYLD and becomes a pro-necrotic signaling molecule. Therefore, non-ubiquitinated RIPK1 can trigger necrotic cell death as well as apoptosis. In fact, we observe that blockade of RIPK1 ubiquitination by expression of the TRAF2DN, mutation of the acceptor lysine (i.e., expression of RIPK1-K377R) or removal of the cIAPs by pharmacological agents greatly potentiates RIPK1-dependent necrosis when caspase activity is blocked. Similarly, NEMO-deficient Jurkat T cells are more sensitive to programmed necrosis upon caspase inhibition than Jurkats rendered NFκB-deficient by over-expression of the IκBαSR. Therefore, we conclude that stable formation of Complex I can prevent RIPK1 from engaging either apoptosis or necrosis. Apoptosis does not require the expression of CYLD, which hints at the possibility that loss of non-degradative ubiquitin chains from RIPK1 enhances apoptosis due to the loss of ubiquitin-binding proteins such as NEMO and that RIPK1 may not require the removal of non-degradative ubiquitin chains in order to bind caspase 8 and trigger apoptosis. In contrast, the removal of ubiquitin chains from RIPK1 by CYLD appears to be a pre-requisite for RIPK1 to trigger necrosis. A more detailed analysis of the structure of RIPK1 in complex with caspase 8 and RIPK3 will be required to determine how non-degradative ubiquitination of RIPK1 impacts the formation of these very different death-signaling complexes.
Caspase 8 cleaves CYLD, both when caspase 8 is functioning as a death-signaling molecule during apoptosis or as a pro-survival signaling molecule. In either scenario, if CYLD is stabilized, necrotic cell death ensues. The importance of the role of CYLD in mediating necrotic cell death in vivo is clear from the studies of intestinal and skin inflammation in conditional FADD knockout mice [86]. As discussed above, chronic inflammation caused by specific knockout of FADD in the intestinal epithelium or skin could be rescued by crossing the mice onto a RIPK3 knockout background. These two studies also demonstrated that loss of the deubiquitinase activity of CYLD results in the abrogation of the chronic inflammatory conditions. In order to confirm that CYLD is the major target for caspase 8 to prevent programmed necrosis in vivo, studies are currently underway to confirm whether or not loss of CYLD can also restore embryonic development and hematopoiesis in caspase 8-deficient mice.
Ligation of the TCR also drives processing of CYLD by caspase 8 (unpublished data), which suggests that caspase 8 and FADD are required during T-cell activation to remove CYLD and prevent necrosis. However, in the TCR signaling pathway, the ability of caspase 8 to remove CYLD is complicated by the fact that the TCR signaling adaptor Malt1 can also proteolytically cleave CYLD [96]. Malt1 is an argase that cleaves CYLD at asparagine 324, generating a stable C-terminal cleavage product that contains the deubiquitinase domain. It remains to be explored how the processing of CYLD by Malt1 affects the ability of CYLD to function as a deubiquitinase toward RIPK1 and regulates programmed necrosis. This brings up the important question of how different caspase 8-containing signaling complexes can regulate CYLD. In the case of TNFR1 pro-survival signaling, caspase 8 forms a complex with cFLIP [83], which prevents caspase 8 from triggering apoptosis but enables caspase 8 to function as an active enzyme toward a restricted range of substrates, such as CYLD. This is inferred from the fact that CYLD is efficiently processed by pro-survival caspase 8 activity in cells that express FLIP. During pro-survival TCR signaling, caspase 8 is also in a complex with cFLIP and Malt1; the activity of Caspase 8 is known to depend on the presence of Malt1 but not Malt1’s proteolytic activity [97]. The role of cFLIP in both these complexes appears to be to keep caspase 8 in an active conformation, but to restrict caspase 8 from processing caspase 3 and initiating apoptosis. We observe that CYLD is efficiently processed by caspase 8 in cells that are undergoing apoptosis and are not anticipated to express cFLIP, for example, the NEMO-deficient cells or cells rendered NFκB-deficient by over-expression of the IκBαSR [95]. Moreover, recombinant caspase 8, which undergoes autoprocessing to the active tetrameric complex characteristic of apoptosis, is also competent to process CYLD with no requirement for cFLIP. Therefore, it appears that activation of caspase 8 in different signaling complexes is sufficient to remove CYLD and maintain RIPK1 in a ubiquitinated form that does not trigger necrotic cell death. cFLIP is one of the main anti-apoptotic factors induced by NFκB activation. cFLIP appears to enable caspase 8 to retain catalytic activity toward a restricted range of substrates, in particular CYLD to prevent necrosis, but avoid the autoprocessing of caspase 8 and subsequent processing of caspase 3 that initiates apoptosis.
In summary, non-degradative ubiquitination of RIPK1 and the stable formation of complex 1 prevent TNF from triggering RIPK1-dependent apoptosis and necrosis. If caspase 8 activity is blocked, the deubiquitinase CYLD is stabilized and can remove these non-degradative ubiquitin chains from RIPK1, which converts the pro-survival Complex I into a death-signaling necrosome. The suppression of RIPK-mediated necrosis by caspase 8 is essential for embryonic development, hematopoiesis and the prevention of chronic inflammation. So when does RIPK1-mediated cell death occur as a beneficial outcome during an immune response?
Programmed necrosis during infection
Stabilization of CYLD can be accomplished by expression of the poxviral caspase inhibitor CrmA, supporting the idea that necrosis may have evolved to permit the removal of virus-infected cells when the virus can block apoptosis. The ability of the necrotic cells to induce inflammation would circumvent the ability of CrmA-expressing virus to block caspase-dependent inflammatory responses. The ability of RIPK1 ubiquitination to control different cell death pathways may also play a role in the immune response to microbial pathogens. Obviously, the activation of NFκB by multiple immune receptors is a target for microbial immune evasion strategies. For example, Shigella encodes the E3 enzyme IpaH9.8, which targets NEMO for degradation in order to block NFκB and prevent an inflammatory response [98]. From our data, we would predict that this also leaves Shigella-infected cells susceptible to RIPK1-mediated cell death, and indeed, epithelial cells are known to undergo necrotic cell death when infected with Shigella [99]. Similarly, E coli can inject a novel type of methyl transferase that specifically methylates the zinc fingers of TAB 2/TAB 3 and prevents them from binding K63-linked polyubiquitin chains [100]. This methylation thus prevents activation of TAK1/IKK downstream of TLR and the TNF receptor. In the case of TNF signaling, we would predict that some cell types in vivo would undergo necrosis as a result of the loss of TAB/TAB 3 TAK1 binding to RIP1. Indeed, the recruitment of TAK1 to ubiquitinated RIPK1 has been reported to prevent RIPK1 from triggering necrosis [101, 102]. Cells appear to have evolved a system to sense the disruption of Complex I (which mediates the NFκB pro-inflammatory signaling pathway) that switches on necrotic cell death in order to act as a backup mechanism that can circumvent these types of immune evasion strategies and trigger an inflammatory response. Although viral modulators of NFκB activation have been identified, it remains to be seen if specific microbial modulators of this pathway could predispose infected cells to undergo programmed necrosis.
As evidenced from this discussion, a complex interplay between ubiquitination, deubiquitination, Caspase 8 activity and NFκB activation is likely to regulate RIPK1-mediated cell death in many different situations in the immune system. This review has focused on the role of NFκB and ubiquitination in the prevention of RIPK1-mediated apoptotic and necrotic death in cells of the immune system. However, the identification of CYLD as a substrate for caspase 8 suggests that regulation of these complex biochemical pathways is likely to be important in other cellular processes such as mitosis, cell migration and wound healing in which CYLD and caspase 8 are known to have diametrically opposing functions.
References
Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–79. doi:10.1146/annurev.iy.12.040194.001041.
Liu JO. The yins of T cell activation. Sci STKE. 2005;2005(265):re1. doi:10.1126/stke.2652005re1.
Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi:10.1146/annurev.immunol.021908.132641.
Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–63. doi:10.1146/annurev.immunol.18.1.621.
Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006(357):re13. doi:10.1126/stke.3572006re13.
Siggers T, Chang AB, Teixeira A, Wong D, Williams KJ, Ahmed B, et al. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-kappaB family DNA binding. Nat Immunol. 2011;13(1):95–102. doi:10.1038/ni.2151.
Smale ST. Hierarchies of NF-kappaB target-gene regulation. Nat Immunol. 2011;12(8):689–94. doi:10.1038/ni.2070.
Laney JD, Hochstrasser M. Substrate targeting in the ubiquitin system. Cell. 1999;97(4):427–30.
Strack P, Caligiuri M, Pelletier M, Boisclair M, Theodoras A, Beer-Romero P, et al. SCF(beta-TRCP) and phosphorylation dependent ubiquitination of I kappa B alpha catalyzed by Ubc3 and Ubc4. Oncogene. 2000;19(31):3529–36. doi:10.1038/sj.onc.1203647.
Yaron A, Hatzubai A, Davis M, Lavon I, Amit S, Manning AM, et al. Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature. 1998;396(6711):590–4. doi:10.1038/25159.
Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274(5288):787–9.
Wang CY, Mayo MW, Baldwin AS Jr. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274(5288):784–7.
Behrends C, Harper JW. Constructing and decoding unconventional ubiquitin chains. Nat Struct Mol Biol. 2011;18(5):520–8. doi:10.1038/nsmb.2066.
Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136(6):1098–109. doi:10.1016/j.cell.2009.03.007.
Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol. 2009;11(2):123–32. doi:10.1038/ncb1821.
Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181–90.
Ting AT, Pimentel-Muinos FX, Seed B. RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996;15(22):6189–96.
Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8(3):297–303.
Lee TH, Shank J, Cusson N, Kelliher MA. The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem. 2004;279(32):33185–91. doi:10.1074/jbc.M404206200.
Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, et al. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7(5):715–25.
Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430(7000):694–9. doi:10.1038/nature02794.
He KL, Ting AT. A20 inhibits tumor necrosis factor (TNF) alpha-induced apoptosis by disrupting recruitment of TRADD and RIP to the TNF receptor 1 complex in Jurkat T cells. Mol Cell Biol. 2002;22(17):6034–45.
Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem. 2006;281(19):13636–43. doi:10.1074/jbc.M600620200.
Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22(2):245–57. doi:10.1016/j.molcel.2006.03.026.
Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected]. Nat Cell Biol. 2006;8(4):398–406. doi:10.1038/ncb1384.
Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, et al. TAB 2 and TAB 3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15(4):535–48. doi:10.1016/j.molcel.2004.08.008.
Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, et al. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134(4):668–78. doi:10.1016/j.cell.2008.07.039.
Windheim M, Stafford M, Peggie M, Cohen P. Interleukin-1 (IL-1) induces the Lys63-linked polyubiquitination of IL-1 receptor-associated kinase 1 to facilitate NEMO binding and the activation of IkappaBalpha kinase. Mol Cell Biol. 2008;28(5):1783–91. doi:10.1128/MCB.02380-06.
Oeckinghaus A, Wegener E, Welteke V, Ferch U, Arslan SC, Ruland J, et al. Malt1 ubiquitination triggers NF-kappaB signaling upon T-cell activation. EMBO J. 2007;26(22):4634–45. doi:10.1038/sj.emboj.7601897.
Wu CJ, Ashwell JD. NEMO recognition of ubiquitinated Bcl10 is required for T cell receptor-mediated NF-kappaB activation. Proc Natl Acad Sci USA. 2008;105(8):3023–8. doi:10.1073/pnas.0712313105.
He KL, Ting AT. Essential role for IKKgamma/NEMO in TCR-induced IL-2 expression in Jurkat T cells. Eur J Immunol. 2003;33(7):1917–24. doi:10.1002/eji.200323650.
Zhang L, Blackwell K, Shi Z, Habelhah H. The RING domain of TRAF2 plays an essential role in the inhibition of TNFalpha-induced cell death but not in the activation of NF-kappaB. J Mol Biol. 2010;396(3):528–39. doi:10.1016/j.jmb.2010.01.008.
Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36(5):831–44. doi:10.1016/j.molcel.2009.10.013.
Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30(6):689–700. doi:10.1016/j.molcel.2008.05.014.
Park SM, Yoon JB, Lee TH. Receptor interacting protein is ubiquitinated by cellular inhibitor of apoptosis proteins (c-IAP1 and c-IAP2) in vitro. FEBS Lett. 2004;566(1–3):151–6. doi:10.1016/j.febslet.2004.04.021.
Vince JE, Pantaki D, Feltham R, Mace PD, Cordier SM, Schmukle AC, et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-{kappa}B and to prevent tnf-induced apoptosis. J Biol Chem. 2009;284(51):35906–15. doi:10.1074/jbc.M109.072256.
Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465(7301):1084–8. doi:10.1038/nature09128.
Luo JL, Kamata H, Karin M. The anti-death machinery in IKK/NF-kappaB signaling. J Clin Immunol. 2005;25(6):541–50. doi:10.1007/s10875-005-8217-6.
Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21(16):5299–305. doi:10.1128/MCB.21.16.5299-5305.2001.
Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1995;81(4):513–23.
Cusson N, Oikemus S, Kilpatrick ED, Cunningham L, Kelliher M. The death domain kinase RIP protects thymocytes from tumor necrosis factor receptor type 2-induced cell death. J Exp Med. 2002;196(1):15–26.
Chan FK, Lenardo MJ. A crucial role for p80 TNF-R2 in amplifying p60 TNF-R1 apoptosis signals in T lymphocytes. Eur J Immunol. 2000;30(2):652–60. doi:10.1002/1521-4141(200002)30:2<652::AID-IMMU652>3.0.CO;2-L.
Pimentel-Muinos FX, Seed B. Regulated commitment of TNF receptor signaling: a molecular switch for death or activation. Immunity. 1999;11(6):783–93.
Natoli G, Costanzo A, Guido F, Moretti F, Bernardo A, Burgio VL, et al. Nuclear factor kB-independent cytoprotective pathways originating at tumor necrosis factor receptor-associated factor 2. J Biol Chem. 1998;273(47):31262–72.
Lee SY, Kaufman DR, Mora AL, Santana A, Boothby M, Choi Y. Stimulus-dependent synergism of the antiapoptotic tumor necrosis factor receptor-associated factor 2 (TRAF2) and nuclear factor kappaB pathways. J Exp Med. 1998;188(7):1381–4.
Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281(5383):1680–3.
Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12(6):633–42.
Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J Biol Chem. 1992;267(25):17971–6.
Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289(5488):2350–4.
Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74(4):597–608.
Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol Cell Biol. 2000;20(8):2687–95.
Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267(5203):1506–10.
Natoli G, Costanzo A, Guido F, Moretti F, Levrero M. Apoptotic, non-apoptotic, and anti-apoptotic pathways of tumor necrosis factor signalling. Biochem Pharmacol. 1998;56(8):915–20.
Li X, Yang Y, Ashwell JD. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature. 2002;416(6878):345–7. doi:10.1038/416345a.
Fotin-Mleczek M, Henkler F, Samel D, Reichwein M, Hausser A, Parmryd I, et al. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J Cell Sci. 2002;115(Pt 13):2757–70.
O’Donnell MA, Legarda D, Skountzos P, Yeh WC, Ting AT. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr Biol. 2007;17(5):418–24. doi:10.1016/j.cub.2007.01.027.
Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133(4):693–703. doi:10.1016/j.cell.2008.03.036.
Legarda-Addison D, Hase H, O’Donnell MA, Ting AT. NEMO/IKKgamma regulates an early NF-kappaB-independent cell-death checkpoint during TNF signaling. Cell Death Differ. 2009;16(9):1279–88. doi:10.1038/cdd.2009.41.
Ch’en IL, Beisner DR, Degterev A, Lynch C, Yuan J, Hoffmann A, et al. Antigen-mediated T cell expansion regulated by parallel pathways of death. Proc Natl Acad Sci USA. 2008;105(45):17463–8. doi:10.1073/pnas.0808043105.
Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in T cells. J Exp Med. 2011;. doi:10.1084/jem.20110251.
Lu JV, Weist BM, van Raam BJ, Marro BS, Nguyen LV, Srinivas P, et al. Complementary roles of Fas-associated death domain (FADD) and receptor interacting protein kinase-3 (RIPK3) in T-cell homeostasis and antiviral immunity. Proc Natl Acad Sci USA. 2011;108(37):15312–7. doi:10.1073/pnas.1102779108.
Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;. doi:10.1038/nature09857.
Vivarelli MS, McDonald D, Miller M, Cusson N, Kelliher M, Geha RS. RIP links TLR4 to Akt and is essential for cell survival in response to LPS stimulation. J Exp Med. 2004;200(3):399–404. doi:10.1084/jem.20040446.
Ma Y, Temkin V, Liu H, Pope RM. NF-kappaB protects macrophages from lipopolysaccharide-induced cell death: the role of caspase 8 and receptor-interacting protein. J Biol Chem. 2005;280(51):41827–34. doi:10.1074/jbc.M510849200.
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1(6):489–95. doi:10.1038/82732.
Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278(51):51613–21. doi:10.1074/jbc.M305633200.
Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140(6):798–804. doi:10.1016/j.cell.2010.02.015.
Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9(5):353–63. doi:10.1038/nri2545.
Li M, Beg AA. Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J Virol. 2000;74(16):7470–7.
Zheng L, Bidere N, Staudt D, Cubre A, Orenstein J, Chan FK, et al. Competitive control of independent programs of tumor necrosis factor receptor-induced cell death by TRADD and RIP1. Mol Cell Biol. 2006;26(9):3505–13. doi:10.1128/MCB.26.9.3505-3513.2006.
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187(9):1477–85.
Kettle S, Alcami A, Khanna A, Ehret R, Jassoy C, Smith GL. Vaccinia virus serpin B13R (SPI-2) inhibits interleukin-1beta-converting enzyme and protects virus-infected cells from TNF- and Fas-mediated apoptosis, but does not prevent IL-1beta-induced fever. J Gen Virol. 1997;78(Pt 3):677–85.
Dobbelstein M, Shenk T. Protection against apoptosis by the vaccinia virus SPI-2 (B13R) gene product. J Virol. 1996;70(9):6479–85.
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–21. doi:10.1038/nature10558.
Ray CA, Black RA, Kronheim SR, Greenstreet TA, Sleath PR, Salvesen GS, et al. Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the interleukin-1 beta converting enzyme. Cell. 1992;69(4):597–604.
Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, Hernanz-Falcon P, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature. 2009;458(7240):899–903. doi:10.1038/nature07750.
Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol. 2008;9(10):1179–88. doi:10.1038/ni.1651.
Caminschi I, Proietto AI, Ahmet F, Kitsoulis S, Shin Teh J, Lo JC, et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood. 2008;112(8):3264–73. doi:10.1182/blood-2008-05-155176.
He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–11. doi:10.1016/j.cell.2009.05.021.
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–6. doi:10.1126/science.1172308.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–23. doi:10.1016/j.cell.2009.05.037.
Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138(2):229–32. doi:10.1016/j.cell.2009.07.006.
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363–7. doi:10.1038/nature09852.
Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471(7338):373–6. doi:10.1038/nature09878.
Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477(7364):335–9. doi:10.1038/nature10400.
Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477(7364):330–4. doi:10.1038/nature10273.
Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, et al. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. 2011;35(4):572–82. doi:10.1016/j.immuni.2011.08.014.
Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135(7):1311–23. doi:10.1016/j.cell.2008.10.044.
Sun SC. Deubiquitylation and regulation of the immune response. Nat Rev Immunol. 2008;8(7):501–11. doi:10.1038/nri2337.
Sun SC. CYLD: a tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2009;17(1):25–34. doi:10.1038/cdd.2009.43.
Wright A, Reiley WW, Chang M, Jin W, Lee AJ, Zhang M, et al. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev Cell. 2007;13(5):705–16. doi:10.1016/j.devcel.2007.09.007.
Tran TM, Temkin V, Shi B, Pagliari L, Daniel S, Ferran C, et al. TNFalpha-induced macrophage death via caspase-dependent and independent pathways. Apoptosis. 2009;14(3):320–32. doi:10.1007/s10495-009-0311-4.
Storz P, Doppler H, Ferran C, Grey ST, Toker A. Functional dichotomy of A20 in apoptotic and necrotic cell death. Biochem J. 2005;387(Pt 1):47–55. doi:10.1042/BJ20041443.
Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The ubiquitin-modifying enzyme A20 is required for termination of toll-like receptor responses. Nat Immunol. 2004;5(10):1052–60. doi:10.1038/ni1110.
O’Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R et al. CASPASE 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. 2011;13:1437–42. doi:10.1038/ncb2362
Staal J, Driege Y, Bekaert T, Demeyer A, Muyllaert D, Van Damme P, et al. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J. 2011;30(9):1742–52. doi:10.1038/emboj.2011.85.
Kawadler H, Gantz MA, Riley JL, Yang X. The paracaspase MALT1 controls caspase-8 activation during lymphocyte proliferation. Mol Cell. 2008;31(3):415–21. doi:10.1016/j.molcel.2008.06.008.
Ashida H, Kim M, Schmidt-Supprian M, Ma A, Ogawa M, Sasakawa C. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF-kappaB-mediated inflammatory response. Nat Cell Biol. 2010;12(1):66–73. doi:10.1038/ncb2006.
Galluzzi L, Kroemer G. Shigella targets the mitochondrial checkpoint of programmed necrosis. Cell Host Microbe. 2009;5(2):107–9. doi:10.1016/j.chom.2009.01.002.
Zhang L, Ding X, Cui J, Xu H, Chen J, Gong YN, et al. Cysteine methylation disrupts ubiquitin-chain sensing in NF-kappaB activation. Nature. 2011;481(7380):204–8. doi:10.1038/nature10690.
Arslan SC, Scheidereit C. The prevalence of TNFalpha-induced necrosis over apoptosis is determined by TAK1-RIP1 interplay. PLoS ONE. 2008;6(10):e26069. doi:10.1371/journal.pone.0026069.
Vanlangenakker N, Vanden Berge N, Berghe T, Bogaert P, Laukens B, Zoberl K, Deshayes K, et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2010;18(4):656–65. doi:10.1038/cdd.2010.138.
Acknowledgments
We thank the current and past laboratory members for insightful discussions throughout the course of studies conducted in the Ting lab. This work was supported by National Institutes of Health grant AI052417 (Adrian T. Ting). Adrian T. Ting is a recipient of the Irma T. Hirschl Career Scientist Award.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
O’Donnell, M.A., Ting, A.T. NFκB and ubiquitination: partners in disarming RIPK1-mediated cell death. Immunol Res 54, 214–226 (2012). https://doi.org/10.1007/s12026-012-8321-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-012-8321-7