
Abstract
Hydrolysis of fatty acyl thioester bonds by thioesterases to produce free fatty acids is important for dictating the diversity of lipid metabolites produced in plants. We have characterized a four-member family of fatty acyl thioesterases from Arabidopsis thaliana, which we have called acyl-lipid thioesterase1 (ALT1), ALT2, ALT3, and ALT4. The ALTs belong to the Hotdog fold superfamily of thioesterases. ALT-like genes are present in diverse plant taxa, including dicots, monocots, lycophytes, and microalgae. The four Arabidopsis ALT genes were found to have distinct gene expression profiles with respect to each other. ALT1 was expressed specifically in stem epidermal cells and flower petals. ALT2 was expressed specifically in root endodermal and peridermal cells as well as in stem lateral organ boundary cells. ALT3 was ubiquitously expressed in aerial and root tissues and at much higher levels than the other ALTs. ALT4 expression was restricted to anthers. All four proteins were localized in plastids via an N-terminal targeting sequence of about 48 amino acids. When expressed in Escherichia coli, the ALT proteins used endogenous fatty acyl–acyl carrier protein substrates to generate fatty acids that varied in chain length (C6–C18), degree of saturation (saturated and monounsaturated), and oxidation state (fully reduced and β-ketofatty acids). Despite their high amino acid sequence identities, each enzyme produced a different profile of lipids in E. coli. The biological roles of these proteins are unknown, but they potentially generate volatile lipid metabolites that have previously not been reported in Arabidopsis.
Similar content being viewed by others


Introduction
Fatty acids are generally activated for metabolism by thioester linkage to either acyl-carrier protein (ACP) or coenzyme A (CoA). The release of free fatty acids from their activating groups by fatty acyl-thioesterases (TEs) is important for regulating lipid metabolism and lipid trafficking. The most widely recognized function of fatty acyl-TEs in plants is to direct newly synthesized fatty acids towards specific metabolic fates. Fatty acid synthesis occurs in the plastids of plants. It is a cyclic pathway that iteratively elongates ACP-linked fatty acids by two carbons units (Li-Beisson et al. 2013). Nascent fatty acids are then partitioned into two cellular locations for further anabolism to produce a wide variety of lipids in the plastid-localized prokaryotic pathway and the endoplasmic reticulum (ER)-localized eukaryotic pathway (Benning 2009). Fatty acyl groups enter the prokaryotic pathway via acyltransferases, which utilize metabolically active fatty acyl-ACPs for plastid membrane biogenesis. Fatty acyl-TEs direct substrates towards the eukaryotic pathway by cleaving the ACP group, thereby releasing metabolically inactive free fatty acids and allowing their export from the plastid.
Fatty acyl-TEs characterized to date mainly belong to one of two fold types: the α/β-hydrolase fold superfamily and the Hotdog fold superfamily (Cantu et al. 2010). Both superfamilies are diverse groups of proteins found in all taxonomical domains. Each fold type is characterized by its tertiary structure, and therefore proteins within the same superfamily can have highly divergent primary amino acid structures. α/β-hydrolase fold proteins are characterized by an eight-stranded β-sheet surrounded by α-helices (Nardini and Dijkstra 1999). This superfamily is one of the largest known, with at least 30,000 known members, with widely ranging enzymatic and non-enzymatic roles (Lenfant et al. 2013). The hallmark of a Hotdog fold domain is a 5–7 stranded antiparallel β-sheet wrapped around a central α-helix. Functionally characterized members of the Hotdog fold protein family have either thioesterase or dehydratase activities (Dillon and Bateman 2004). Hotdog fold domains dimerize or form higher order oligomers in vivo, although there are examples of a single polypeptide containing two Hotdog fold domains (Pidugu et al. 2009).
There are two types of highly conserved plant acyl-ACP TEs described to date that terminate fatty acid synthesis via hydrolysis of acyl-ACP thioester bonds: FATA- and FATB-type TEs. Both types are plastid localized enzymes with double Hotdog fold domains. FATA-type acyl-TEs are highly active towards monounsaturated acyl-ACP substrates, oleoyl-ACP (18:1Δ9) in particular, while FATB-type acyl-TEs act preferentially on saturated acyl-ACPs (Jones et al. 1995). In most plant tissues, palmitic acid (16:0) is the major saturated fatty acid produced, although alternate FATB enzymes are expressed in seeds of certain plants (Voelker et al. 1992). Together, FATA and FATB TEs direct fatty acid precursors to the ER for generation of various lipid classes including phospholipids, triacylglycerols and sphingolipids, as well as specialized lipids such as cuticular waxes, cutin, and suberin.
In specific cases, highly specialized roles for plant fatty acyl-TEs have been demonstrated or proposed. The alternate FATB enzymes described above have a preference for saturated fatty acyl-ACPs of medium chain lengths (C8–C14), leading to differing seed oil profiles between species (Voelker et al. 1992). In wild tomato, Solanum habrocaites subspecies glabratum, a fatty acyl-ACP TE called methylketone synthase2 (ShMKS2) acts on C12–C16 β-ketoacyl-ACPs, which are intermediates of the fatty acid synthesis cycle (Ben-Israel et al. 2009; Yu et al. 2010). The resulting β-ketofatty acids are then converted to methylketones by the decarboxylase enzyme ShMKS1 (Auldridge et al. 2012). These methylketones accumulate in trichomes and are insecticides (Fridman et al. 2005). Although ShMKS2 is a single Hotdog fold protein, it is not similar at the primary amino acid level to the FATA or FATB double Hotdog fold proteins. Synthesis of cannabinoids by Cannabis sativa begins with a 6:0 fatty acid, which may be generated by a fatty acyl-ACP TE that interrupts fatty acid synthesis (Fellermeier et al. 2001; Marks et al. 2009), although no such enzyme has yet been identified. A portion of cuticular waxes on aerial surfaces are often very-long chain (e.g. 22:0–30:0) free fatty acids (Bernard and Joubès 2012), presumably generated by an ER-localized very-long-chain fatty acyl-CoA TE (Lü et al. 2009). Fatty acyl-CoA TEs have also been implicated in peroxisomal oxidation of fatty acids in mammals (Hunt et al. 2012), which may apply to plants as well.
Four Arabidopsis thaliana fatty acyl-TEs have been experimentally characterized to date: two redundant FATA-type acyl-ACP TEs, one FATB-type acyl-ACP TE, and an acyl-CoA TE called ACH2. A fata1 fata2 double mutant in which the expression of each gene is reduced by approximately 50 % has a modest decrease in lipid pools, most pronounced in seed triacylglycerols, and with a particular deficiency in 18:1 and 18:2 fatty acids (Moreno-Pérez et al. 2012). A nearly complete knockdown mutant of FATB has a 20–50 % reduction in saturated components of eukaryotic pathway derived products, accompanied by a dwarf phenotype and reduced viability (Bonaventure et al. 2003). ACH2 is a peroxisomal acyl-CoA TE with in vitro activity towards 16:0-CoA, but its biological role is currently unknown (Tilton et al. 2004).
Considering the diversity of plant lipid metabolic pathways, it is highly likely that Arabidopsis expresses many additional fatty acyl-TEs. Indeed, the Arabidopsis genome encodes several genes annotated as potential acyl-TEs. We report here on the characterization of a four member Arabidopsis gene family that we have called ACYL-LIPID THIOESTERASE1 (ALT1), ALT2, ALT3, and ALT4, which encode single Hotdog fold fatty acyl-ACP TEs related to wild tomato ShMKS2.
Materials and methods
Quantitative real-time PCR
Tissues were harvested from Col-0 wild-type plants, flash frozen in liquid N2, and stored at −80 °C. Most tissues (stems, rosette leaves, cauline leaves, flowers, and siliques) were harvested from 6-week-old plants, grown in long-day conditions (18 h light/6 h dark). Roots and whole seedlings were harvested after 15 days of growth on solid Murashige and Skoog (MS) media (no sucrose) and grown under continuous light. RNA was extracted using a commercial kit and DNase treated (RNaqueous kit and Turbo DNase, Applied Biosystems, Carlsbad, CA, USA), following the manufacturer’s instructions. RNA isolation aid (Applied Biosystems) was used during preparation of RNA from carbohydrate-rich silique tissues. cDNA was synthesized from 1 μg of total RNA with Superscript-reverse transcriptase III (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Quantitative PCR was performed using a StepOnePlus instrument and PowerSyber qPCR mastermix (both from Applied Biosystems). Each 10 μl reaction contained 1× PowerSyber mix, 50 nM of each primer (Supplementary Table S1), and cDNA template (1 % of the reverse transcriptase reaction). Transcripts were quantified using absolute standard curves, which were generated with a 10-fold dilution series using 10−5–1 pg of plasmid DNA as template. These plasmids were generated by cloning either open reading frames plus 3′ untranslated regions (for ALT genes) or the qPCR amplicon only (endogenous control genes) into pCR-Blunt II-TOPO (Invitrogen, Carlsbad, CA, USA). ALT transcripts were normalized with the endogenous controls ACT2 (At1g49240), eIF4A-1 (At3g13920), and GAPC (At3g04120). The transcript abundance of the three endogenous control genes were averaged, and ALT1–4 expression is reported relative to this value. Each tissue had 2–3 biological replications, and every reaction was done as 3 technical replications. Due to the high level of sequence similarity between ALT1–4, we tested the specificity of qPCR primers using cloned transcripts as templates. After testing every primer and template combination, the highest level of cross hybridization was obtained with ALT1 primers towards the ALT2 template. This reaction had a Ct value of 11.7 cycles above the reaction of ALT1 primers towards the ALT1 template. From this, we conclude that no more than 0.03 % (1/211.7) of the amplification of a given target is due to cross hybridization of primers to different ALT genes.
Promoter::GUS analysis
Promoter sequences were amplified by PCR using bacterial artificial chromosome plasmids as templates (T9I1 for ALT1 and ALT2; T22E19 for ALT4), with the primers ALT<1–4>-Prom-For-SalI and ALT<1–4>-Prom-Rev-BamHI (Supplementary Table S1). These fragments were inserted into pBI101.1 (Clontech, Mountain View, CA, USA) between the SalI and BamHI restriction sites and PCR amplified regions were verified by DNA sequencing. The resultant constructs contained ~1.8 kb of upstream sequence fused to the β-glucuronidase reporter gene. These constructs were transformed into Agrobacterium tumefaciens strain GV3101::pMP90 by electroporation. These Agrobacteria were used to transform Arabidopsis by the floral dip method (Clough and Bent 1998). Tissues were prepared for histochemical staining by soaking tissues in heptane for 30 s, air drying for 1 min, and then rinsing with GUS staining buffer (50 mM NaH2PO4, 100 mM KFe(CN)6, 0.1 % Triton X-100). Staining was performed by incubation at 37 °C for at least 2 h and up to overnight in GUS staining buffer with 2 mM 5-bromo-4-chloro-indolyl-β-d-glucuronic acid (X-Gluc). Whole tissues were cleared by replacing the buffer with 70 % ethanol and incubating at room temperature overnight, and then imaged with a Zeiss SteREO Discovery V20 stereomicroscope (Carl Zeiss MicroImaging, Inc., Thornwood, NY, USA).
Root cross sections were made from 10-day-old roots embedded in LR white resin (London Resin Company Ltd., London, UK) or from 5-week-old roots embedded in paraffin (Paraplast plus, Sigma-Aldrich, St. Louis, MO, USA) for examination of endodermal and peridermal cell layers, respectively. These sections were imaged with a Zeiss Axio Imager M2 compound microscope. To embed in LR white resin, roots were removed from the X-Gluc solution, soaked in 50 % ethanol for 1 h, and then fixed overnight in 25 mM NaPO4 (pH 6.8) + 2 % glutaraldehyde at 4 °C. Samples were dehydrated with an ethanol series (50, 60, 70, 80, 90, 100 %) for 2 times 30 min each, embedded in resin following the manufacturer’s recommendations, cut to 10 μm sections with an ultra-microtome, and then imaged. To embed tissue in paraffin, tissues were fixed in FAA solution (3.7 % formaldehyde, 50 % ethanol, 5 % acetic acid) for 3 h, dehydrated with an ethanol series (70, 80, 90, 95 % for 30 min each), and then left in 100 % ethanol overnight. Tissues were infiltrated by incubation in 25 % tert-butanol in ethanol for 30 min, followed by 50 % tert-butanol in ethanol for 30 min, then 100 % tert-butanol overnight at 60 °C, overnight in 50 % paraffin in tert-butanol at 60 °C, and finally in 100 % paraffin at 60 °C for 3 days, changing the paraffin twice a day. The paraffin was allowed to solidify at 4 °C for a few hours, and then cut into 10 μm sections with a microtome. Sections were fixed to a glass slide at 42 °C overnight, dewaxed with tert-butanol, and then imaged.
In situ hybridization
An antisense probe specific for ALT1 was amplified from the clone U14921 (obtained from the Arabidopsis Biological Resource Centre at Ohio State University) containing the cDNA sequence for ALT1 using the primers ALT1_Antisense_Fwd and ALT1_Antisense_Rev (Supplementary Table S1) that appends the T7 polymerase binding site to the 5′ end of the template strand. As a negative control, a sense probe was also amplified from the same clone using the primers ALT1_sense_Fwd and ALT1_sense_Rev (Supplementary Table S1). Probe synthesis was performed as described previously (Hooker et al. 2002). Stem tissue for in situ hybridization was harvested from 6-week-old wild-type plants and fixed in formal-acetic-alcohol (50 % ethanol, 5 % acetic acid, 3.7 % paraformaldehyde). Tissues were embedded in paraffin as described previously (Samach et al. 1997) and 8 μm cross sections of stem were sectioned with a rotary microtome and placed on frosted slides (Superfrost Plus, Fisher Scientific). The sections were bonded to the slides by incubating the slides at 42 °C overnight on a slide warmer. Slide preparations were de-waxed and hybridized with probe as described previously (Samach et al. 1997). The hybridized sections were imaged under 50 % glycerol using a Zeiss Axio Imager M2 compound microscope.
Subcellular localization
Full length ALT1 and ALT2 open reading frames were amplified by PCR with the primers ALT<1–2>-ORF-For-XbaI and ALT<1–2>-ORF-RevNoStop-BamHI. Full length ALT3 and ALT4 open reading frames were amplified with the primers ALT<3–4>-ORF-For-BamHI and ALT<3–4>-ORF-RevNoStop-BamHI. The open reading frame for ALT11–47 was amplified with ALT1-ORF-For-XbaI and ALT1-ORF-Trunc-RevNoStop-BamHI, and the open reading frame for ALT148–189 was amplified with ALT1-ORF-Trunc-For-XbaI and ALT1-ORF-RevNoStop-BamHI (Supplementary Table S1). These fragments were ligated into pVKH18-35S::GFPC (Dean et al. 2007), and verified by DNA sequencing. The resultant constructs produced 35S promoter driven, C-terminal GFP fusion proteins (ALT–GFP). The constructs were transformed into Agrobacteria as described above, and the resultant strains were infiltrated into leaves of Nicotiana benthamiana as described previously (Sparkes et al. 2006). GFP fused to the targeting peptides of Nicotiana tabacum rubisco small subunit and Saccharomyces cerevisiae cytochrome c oxidase IV, were used as localization markers for plastids and mitochondria, respectively, as described previously (Nelson et al. 2007). Three days post infiltration, leaves were imaged by confocal microscopy with a Zeiss LSM 510 Meta using a Plan Apochromat 63x objective (Carl Zeiss MicroImaging, Inc., Thornwood, NY). Samples were excited with a 488 nm argon laser and emission was recorded between 505 and 565 nm for GFP and LP 650 nm for chlorophyll autofluorescence.
Expression in E. coli
The coding sequences for ALT1–4 proteins lacking their N-terminal plastid targeting sequences were amplified using ALT<1–4>-ORF-Trunc-For-BamHI and ALT<1–4>-ORF-Rev-EcoRI primers (Supplementary Table S1), and inserted into pET-28a (EMD4Biosciences, Gibbstown, NJ, USA) using the indicated restriction sites. The coding sequence for a truncated FATB lacking the plastid targeting sequence (Mayer and Shanklin 2007) was amplified using FATB-ORF-trunc-For-SacI and FATB-ORF-Rev-HindIII, and ligated into the corresponding sites of pET28a. These constructs were transformed in E. coli K27, in which the long-chain acyl CoA synthetase gene, FadD, had been knocked out and the bacterial strain also modified for inducible expression of T7 RNA polymerase (Lü et al. 2009). E. coli strains were grown in Lysogeny broth (LB)-Miller at 37 °C to A 600 = 0.6, then protein expression was induced by the addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside and the cells grown for 16–18 h at 18 °C. Bacterial cells were pelleted by centrifugation at 4,500×g for 5 min in glass vials. One ml of the supernatant was mixed with 1 ml 20 mM H2SO4 and incubated for 30 min at 75 °C, then 30 min at 30 °C. 50 μg tetracosane (24:0 alkane) was added as an internal standard, and then lipids were extracted into 500 μl hexane. One μl of the hexane fraction was used for gas chromatographic analysis. Lipids were quantified with a Varian GC3900 equipped with a HP-5MS column (30 m length, 0.25 mm inner diameter, 0.25 μm film thickness) and a flame ionization detector. Samples were injected in splitless mode. The carrier gas was helium with a constant flow rate of 2 ml/min. The column oven was initially at 50 °C for 8 min, then ramped at 15 °C/min up to 275 °C, which was held for 5 min, giving a total run time of 28 min. Identification of novel products was performed using an Agilent 6850 gas chromatograph equipped with an HP-5MS column (30 m length, 0.25 mm inner diameter, 0.25 μm film thickness) and an Agilent 5975 mass spectrometric detector (70 eV, mass-to-charge ratio 50–750). The initial oven temperature of 33 °C was held for 5 min, then ramped at 2 °C/min up to 65 °C, which was held for 2 min, then ramped at 50 °C/min up to 320 °C, which was held for 1.9 min, giving a total run time of 30 min. The carrier gas was helium with a constant flow rate of 1.5 ml/min.
Identification of novel compounds by mass spectrometry
All saturated FAs and MKs were identified by comparison of experimental spectra to those of commercial standards. Unsaturated FAs were also identified by comparison to standards, although the position of the double bond was not investigated, but rather assumed based on the typical E. coli lipid profile. 11:1 and 13:1 MKs were identified by comparison to the spectra provided in Goh et al. (2012), and 15:1 MK was identified by the presence of the calculated molecular ion and the characteristic MK fragments (Supplementary Fig. 3S), as well as the retention patterns of saturated versus unsaturated MKs.
In vitro thioesterase assay
The ALT1 catalytic mutant was made by site directed mutagenesis using overlap extension PCR. The pET28a::ALT1 clone described above was used as the template and the primers used were ALT1-ORF-Trunc-For-BamHI, ALT1-ORF-Rev-EcoRI, ALT1-mutant-DA-For, and ALT1-mutant-DA-Rev (Supplementary Table S1). This converted the aspartate residue at position 64 (of the full length protein) to alanine. This clone was transformed into the E. coli expression strain described above.
Bacterial strains carrying the wild-type and mutant ALT-containing plasmids were inoculated from overnight cultures into 250 ml of terrific broth (12 g/L tryptone, 24 g/L yeast extract, 0.4 % glycerol, 89 mM K-phosphate buffer pH 6.2) and grown to A 600 = 0.4 and then protein expression was induced with 0.3 mM IPTG and cells were cultured at 10 °C for approximately 40 h. Cells were harvested by centrifugation, washed with phosphate buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4), and stored at −80 °C until use. Proteins were purified using PrepEase Histidine-Tagged Protein Purification Midi Columns (Affymetrix, Santa Clara, CA, USA). Cell pellets resuspended in 5 ml lysis-binding buffer (50 mM Na phosphate, 300 mM NaCl, 10 mM imidazole, 0.5 % Triton X-100, pH 8.0), and lysed by sonication. Lysates were clarified by centrifugation, and then loaded onto the commercial columns. Columns were washed with 3× 2 ml lysis-binding buffer, and then histidine-tagged proteins eluted with 5 ml elution buffer (same composition as lysis-binding buffer, but with imidazole concentration raised to 250 mM). Eluates were dialyzed overnight against 5 l dialysis buffer (50 mM Na phosphate, 500 mM NaCl, 1 M (NH4)2SO4, 2 mM DTT, 15 % glycerol, pH 6.8), divided into single use aliquots, flash frozen in liquid N2, and stored at −80 °C until use. Total protein concentrations were determined using the Bio-Rad protein assay kit (Bio-Rad, Hercules, CA, USA), using bovine serum albumin as a standard. Thioesterase concentrations were determined using Western blotting for the T7 epitope by comparing signal intensity to a commercial standard of known concentration. Purity of the ATL1, ALT1-D64A, and ALT2 preparations ranged from 10 to 12 %, similar to that obtained previously for ShMKS2 (Auldridge et al. 2012). The ALT3 and ALT4 preparations had notably higher purity, 39 and 21 %, respectively.
The 14C labeled 12:0-ACP substrate, prepared with recombinant spinach ACP, was generously provided by John Shanklin (Biosciences Department, Brookhaven National Laboratory). Assays were set up in 100 μl reactions containing 25 mM Na phosphate buffer, pH 8.0, 50 mM KCl, and 10 μl thioesterase preparation (diluted to about 40 ng TE/μl in dialysis buffer). The reaction was initiated by addition of 30 pmol acyl-ACP substrate, incubated at room temperature for 30 min, and then terminated by addition of 500 μl of 1 M acetic acid in isopropanol. Free fatty acids were extracted 2× with 500 μl hexane, pooled, and quantified by liquid scintillation counting. Control reactions in which 12:0-ACP substrate was extracted directly, without incubation, gave a small background hydrolysis value that was subtracted from all the other reaction calculations.
Results
ALT1–4 form a highly related Arabidopsis gene family with homologues in many plant taxa
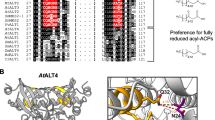
The ALT1–4 genes each consist of 5 exons and 4 introns and are present in pairs at two genomic loci on chromosome 1. ALT1 (At1g35290) and ALT2 (At1g35250) are separated by four genes, while ALT3 (At1g68260) and ALT4 (At1g68280) are separated by a single gene. The four ALT genes encode small proteins (188–192 amino acids) that are very similar to one another, sharing 66–83 % amino acid sequence identity (76–88 % similarity). These predicted proteins are 72–80 % similar (50–62 % identical) to wild tomato ShMKS2. A multiple sequence alignment between ALT1–4 and ShMKS2 illustrates the high sequence similarity between these proteins, especially in the C-terminal ~140 amino acids that corresponds to a single Hotdog fold domain (Fig. 1). The amino acid sequence similarity of ALT1–4 in the Hotdog fold domain alone is 82–91 % (72–89 % identity). Although the plant protein sequences are very different from the bacterial enzyme, the experimentally characterized active site residues of the Escherichia coli TE, YbgC, (aspartate, glycine, and valine in the DXXGXV motif) are conserved in the five plant proteins (Fig. 1) (Benning et al. 1998; Yu et al. 2010). Each ALT protein contains a predicted plastid targeting sequence at the N-terminus. The N-terminal plastid targeting sequences are likely cleaved upon delivery of the proteins to their destination, as this is common for other plastid proteins.

Amino acid sequence alignment of ALT1–4 from Arabidopsis thaliana (At), MKS2 from the wild tomato species Solanum habrochaitis subspecies glabratum (Sh), and YbgC from Escherichia coli (Ec). The predicted plastid targeting sequences of the plant proteins are indicated with a dashed box. Each of the proteins contains a single Hotdog fold domain encompassing the rest of the predicted protein sequence. The experimentally characterized active site residues of the bacterial protein are marked with arrows. These active site residues are conserved in the plant proteins
A search of various genomes identified potential ALT gene homologues in a wide variety of plant species, but none outside the Plantae kingdom. These species include monocots, dicots, the lycophyte Selaginella moellendorffii, and the green microalgae Chlamydomonas reinhardtii and Coccomyxa subellipsoidea. The moss Physcomitrella patens does not contain an ALT homologue. None of the non-plant genomes searched contained homologues. The only ALT protein homologue to be characterized experimentally is ShMKS2. Most plant genomes contain 4–5 homologues, although several contain only a single homologue (e.g. Brassica rapa, Glycine max, Poplus trichocarpa). The well characterized fatty acyl-ACP TEs FATA and FATB contain two sequential Hotdog fold domains, but these proteins are otherwise not similar to the ALTs. In a phylogenetic tree, the ALTs proteins form a distinct clade from FATA and FATB proteins (Supplementary Fig. S1).
ALT1–4 have distinct gene expression patterns
The gene expression patterns of ALT1–4 were examined by four complementary methods: analysis of publically available DNA microarray data, quantitative reverse transcriptase-PCR (qPCR), in situ transcript hybridization, and histochemical staining of promoter::GUS lines.
Examination of tissue-specific gene expression by qPCR revealed ALT1 transcript levels were highest in stem and floral organs and at lower levels in siliques, whole seedlings, and cauline leaves (Fig. 2). ALT1 transcripts were not detected in rosette leaves or in roots. Histochemical staining of ALT1 promoter::GUS reporter lines confirmed expression in stems (Fig. 3a). An in situ hybridization experiment showed that the ALT1 transcript was restricted to epidermal cells in the stem (Fig. 3b, c). There was GUS reporter gene expression in the top portion of the gynoecium (fused carpels) immediately beneath the stigma and in the petals (Fig. 3d). These promoter::GUS lines also showed staining throughout the silique, with strongest staining at the abscission zone (Fig. 3e). In cauline leaves, staining was observed in trichomes. When probing publically available DNA microarray data for genes that co-regulate with ALT1, we noted that several of the top ranked genes are involved in the biosynthesis of cuticular waxes by epidermal cells. These genes included MAH1, CER4, WSD1, CER1, and CER3 (Table 1).

Tissue-specific gene expression patterns of Arabidopsis ALT1–4 analyzed by quantitative reverse transcriptase-PCR. Transcript levels were quantified with absolute standard curves, and normalized with expression of the endogenous control genes ACT2, eIFA4a, and GAPC

Expression patterns of ALT1 (a–f), ALT2 (g–m) and ALT4 (n–p) examined by promoter:: GUS analysis (a, d–p) and in situ hybridization (b, c). a stem; b c stem cross section; d flower; e silique; f rosette leaf; g young root; h mature root; i boundary of shoot and petioles; j silique; k young root cross section; l mature root cross section; m shoot/petiole boundary cross section with arrows pointing to stained cells; n flower cluster; o stamen; p anther cross section. Scale bars: 2 mm (a, e, f), 1 mm (g, h, i, j, n), 0.5 mm (d), 0.25 mm (b, o), 0.1 mm (c, p), 50 μm (l), 25 μm (k, m)
ALT2 was highly expressed in root and stem tissues, with lower expression in siliques and whole seedlings, as determined by tissue-specific qPCR (Fig. 2). The magnitude of ALT2 expression in qPCR experiments was comparable to that of ALT1. Staining of ALT2 promoter::GUS roots was not continuous along the length of the root, but instead had a patchy pattern (Fig. 3g, h). ALT2 expression was limited to endodermal and peridermal cells in young and mature roots, respectively, as revealed in cross sections of stained roots (Fig. 3k, l). GUS staining of siliques showed ALT2 expression in the developing seeds, but not in the silique valves (Fig. 3j). The stem expression of ALT2 was localized to lateral organ boundaries (Fig. 3i), and not along the stem length as was the case with ALT1. A cross section of a stem/petiole boundary showed darker GUS staining throughout all cell layers of the petiole, and lighter staining in the shoot, centered around the vascular cells (Fig. 3m). In silico analysis of DNA microarray experiments identified several suberin biosynthetic genes as being co-expressed with ALT2. These included FAR4, CYP86A1/HORST, CYP86B1/RALPH, ASFT, and GPAT5 (Table 1).
The tissue-specific qPCR experiments showed that ALT3 was expressed in every tissue examined, both aerial and root (Fig. 2). The transcript levels were much higher than ALT1, ALT2, or ALT4. These results are consistent with data from publically available DNA microarray experiments. Histochemical staining of ALT3 promoter::GUS transgenic lines gave very different results between lines, and thus no informative data on the cell-specific expression pattern of ALT3 could be generated using promoter–reporter gene fusions. It is possible that cis-acting DNA elements outside of the upstream ~1.8 kbp region are important for conferring its gene expression pattern. No known lipid metabolic genes were found to have a high co-expression correlation with ALT3, likely due to its ubiquitous expression.
ALT4 was expressed in flowers, but the level of transcript was several orders of magnitude lower than the other genes tested as determined by qPCR (Fig. 2). GUS staining showed specific expression in anthers of flowers (Fig. 3n, o). An anther cross section revealed staining both in anther walls (endothecium) and in microspores (Fig. 3p). This highly restricted expression of ALT4 could explain the very low transcript abundance within the whole flower clusters used for our qPCR experiment. Co-expression analysis was not possible for ALT4 because it is not represented on the 22 K ATH1 gene chip.
ALT proteins localize to plastids
Subcellular localization prediction algorithms (Emanuelsson et al. 2000; Nielsen et al. 1997) indicated that ALT1–4 proteins contain N-terminal plastid targeting sequences of 47–51 residues. Although plastid localization was most likely for all four proteins, ALT2 and ALT4 also had high scores for mitochondrial targeting. We determined the subcellular localization of ALT1–4 by Agrobacterium-mediated transient expression of C-terminal green fluorescent protein (GFP) fusions (ALT–GFP) in leaf cells of Nicotiana benthamiana using confocal microscopy. GFP fusions of full-length ALT proteins all produced green fluorescent signal that co-localized with red plastid autofluorescence (Fig. 4c–f), indicating plastid localization. The same fluorescence pattern was seen when the first 47 amino acids of ALT1 were fused to GFP, whereas a truncated ALT1 lacking these 47 residues was not plastid localized (Fig. 4a, b). This indicates that the plastid targeting sequence of ALT1 is necessary and sufficient for plastid targeting of this protein, which is likely also the case for ALT2, ALT3, and ALT4.

Subcellular localization of ALT-GFP fusions expressed transiently in leaves of Nicotiana benthamiana and observed with confocal microscopy. Fusions of full-length ALT1–4 were analyzed (c–f), as well as a fusion of only the predicted chloroplast targeting signal (CTP) of ALT1 (a) or a version of ALT1 lacking the predicted targeting signal (ΔCTP) (b). GFP signal is shown in green and plastid autofluoresence is in red. Plastid and mitochondrial organelle markers used were described previously (Nelson et al. 2007). The scale bar in panel h represents 50 μm and all panels are the same magnification
ALT1–4 generate fatty acids and β-ketofatty acids when expressed in E. coli
We characterized the fatty acyl-TE activities of ALT1–4 by heterologous expression in E. coli using endogenous acyl-ACP substrates and analyzing lipid products secreted into the media. For these experiments, we used the E. coli mutant fadD, which lacks acyl-CoA synthetase activity (Overath et al. 1969), causing free fatty acids to accumulate and get exported to the media, rather than being activated and degraded by β-oxidation. Due to the high sequence similarity between ALT1–4 and ShMKS2, we analyzed not only unmodified fatty acids as potential enzymatic products, but also β-ketofatty acids. Since β-ketofatty acids are difficult to detect directly, they were chemically decarboxylated to methylketones prior to extraction, as was done by Yu et al. (2010). The methylketones produced by this procedure are shortened by one carbon relative to the initial β-ketofatty acid.
Analysis of the medium of E. coli containing empty vector (negative control) revealed small amounts of saturated and unsaturated fatty acids ranging from 8 to 18 carbons (the most being 16:1 with 127.1 ng/OD unit) as well as traces of 13:0 and 15:1 methyl ketones (44.4 and 48.2 ng/OD unit, respectively) (Figs. 5, 6). All heterologous proteins were successfully expressed and accumulated to similar levels, as observed by Western blotting (Supplementary Fig. S2). As a positive control, we expressed the well characterized Arabidopsis fatty acyl-ACP thioesterase FATB. Expression of FATB in E. coli resulted in large amounts of 16:1 fatty acid (5481.5 ng/OD unit), as previously reported (Mayer and Shanklin 2007). Expression of ALT1–4 each resulted in the production of fatty acids and/or β-ketofatty acids (the latter converted to methylketones for GC analysis) that were either absent or much less abundant in the empty vector negative control (Figs. 5, 6). The structures of all these products were confirmed by GC–MS analyses (Supplemental Fig. S3). The major products of the ALT1-expressing E. coli strain was 12:0 (1250.3 ng/OD unit) with medium-to-long chain fatty acids 14:1, 14:0, 16:1, and 16:0 also being prominent (Figs. 5, 6). ALT2 expression resulted in high levels of medium chain methylketones 7:0 (1,440.5 ng/OD unit) and 9:0 (445.3 ng/OD unit) and smaller levels of 8:0 fatty acid (Figs. 5, 6). ALT3 expression generated predominantly 13:1 methylketone (891.5 ng/OD unit) along with smaller amounts of 7:0, 9:0, 11:0, 11:1, 13:0, and 15:0 methylketones. Fatty acid species were also produced by heterologous expression of ALT3, the most prominent fatty acid being 16:1 (448.2 ng/OD unit) (Figs. 5, 6). Finally, the ALT4-expressing strain generated high amounts of 6:0 (741.1 ng/OD unit) and 8:0 (1,429.0 ng/OD unit) medium chain fatty acids as well as small increases of 10:0 and 16:1 fatty acids and small amounts of 7:0, 13:0 and 15:1 methylketones (Figs. 5, 6). For all proteins tested, both fatty acids and methylketones were generated, but with one of the two chemical classes usually dominating. Compounds of the non-dominant chemical class mirrored the relative levels of the corresponding products in the dominant class. For example, FATB produced mainly 16:1 and 14:0 fatty acids, with much smaller amounts of 15:1 and 13:0 methylketones, while ALT2 generated large amounts 7:0 methylketones, alongside smaller amounts of 8:0 fatty acids. The major products of ALT3 were 13:1, 11:0, and 13:0 methylketones, mirrored by 14:1, 12:0 and 14:0 fatty acids. ALT3 also produced notable amounts of 16:1 fatty acid (mirrored by a small amount of 15:1 methylketone), making it the only protein tested to have main products in both the fatty acid and methylketone classes.

Gas chromatograph traces of lipids secreted into the media of E. coli cultures carrying an empty vector, or expressing ALT1–4, or FATB. The β-ketofatty acids produced were chemically decarboxylated by heat and acid for conversion to more stable methylketones prior to extraction and analysis by gas chromatography. Lipids were identified by GC–MS (see Supplementary Fig. S3) and are indicated above the traces (FA fatty acid, MK methylketone)

Quantification of lipids secreted into the media of E. coli cultures carrying an empty vector, or expressing ALT1–4, or FATB. a fatty acids detected, b methylketones detected, which were derived from β-ketofatty acids. The β-ketofatty acids produced by thioesterase activities were chemically decarboxylated by heat and acid for conversion to more stable methylketones prior to extraction and analysis by gas chromatography. Quantification was done by comparison of peak areas to that of a tetracosane internal standard using GC–FID
In this experimental system, we were not able to observe β-ketofatty acids directly but rather assumed their presence based on detection of methylketones. To ensure that the methylketones detected were in fact chemically derived, rather than direct enzymatic products, extractions were performed without the heat and acid treatment used for decarboxylation of β-ketofatty acids. For this experiment, ALT3 was chosen as it generates both fatty acids and methylketones, as well as FATB because fatty acids are its only known biologically relevant products. As expected, this omission reduced the production of all methylketone species considerably, with no apparent correlation to chain length (75–95 % reduction; Supplementary Fig. S4).
ALT1, 3, and 4 have lauroyl (12:0)-ACP thioesterase activity in vitro
To further examine the enzymatic activities of ALT1–4, we prepared recombinant ALT proteins lacking plastid targeting sequences (as described above) and assayed in vitro their activities towards 12:0-ACP (Fig. 7). A background level of 12:0-ACP hydrolysis was observed in the absence of added protein (buffer alone). Inclusion of recombinant ALT1 increased the level of acyl-ACP hydrolysis above background levels (statistically significant, p < 0.005). To further evaluate ALT1 activity and to account for potential co-eluting E. coli proteins contributing to acyl-ACP hydrolysis, we engineered a catalytically inactive ALT1 mutant to act as another negative control. This mutant was generated by replacing within the active site a conserved aspartate residue (position 64) to alanine (Fig. 1). The corresponding residue in the Pseudomonas sp. enzyme, 4-hydroxybenzoyl-CoA thioesterase, was identified as an active site residue by its proximity to the thioester carbonyl group in the crystal structure and its conservation in other known TEs (Benning et al. 1998). In agreement, the analogous aspartate-to-alanine substitution abolished ShMKS2 thioesterase activity (Yu et al. 2010). This ALT1 catalytically inactive mutant, ALT1-D64A, generated no fatty acids or β-ketofatty acids when expressed in the E. coli fadD expression strain (data not shown). In the in vitro thioesterase assay using 12:0-ACP substrate, ALT1-D64A did not increase hydrolysis above background levels. ALT3 and ALT4 preparations had relatively high 12:0-ACP thioesterase activities, while the ALT2 preparation had no activity above background (Fig. 7).

In vitro 12:0-ACP TE activities of recombinant ALT1–4 and catalytically inactive ALT1-D64A. 400 ng of each TE was incubated with 30 pmol 14C-labeled 12:0-ACP at room temperature for 30 min. Hydrolyzed 12:0 fatty acid (free acid) was extracted with hexane and amounts determined by liquid scintillation counting. Errors bars are ±SD, n = 3–5). A student’s t test comparing hydrolysis levels of ALT-containing reactions to the buffer only control gave p values of <0.005 (*) for ALT1 or <5 × 10−7 (**) for ALT3 and ALT4
Discussion
ALT1–4 encode fatty acyl-ACP TEs with varied substrate preferences
The ALT gene family encodes four Hotdog fold proteins that are highly related to each other, as well as to ShMKS2, for which β-ketoacyl-ACP TE activity has been demonstrated in vitro (Yu et al. 2010). To investigate the hypothesis that ALT1, 2, 3, and 4 are acyl-ACP TEs, we expressed these four proteins in the E. coli mutant fadD. Monitoring acyl-ACP TE activity is often performed in this bacterial expression system, which has abundant acyl-ACP substrates, like the plastid where these enzymes are localized in planta. Acyl-ACP TEs previously tested in such experiments generate products that closely match their known or suspected biological products, both in terms of oxidation state and chain length (Dörmann et al. 1995; Yu et al. 2010). However, discrepancies are sometimes seen with regard to the saturation level of the fatty acids produced in these experiments. For example, the in planta product of FATB is known to be 16:0 fatty acid, but studies that measured fatty acids accumulating in FATB-expressing E. coli gave differing results. In one instance, 16:0 was the predominant product in E. coli (Dörmann et al. 1995), whereas in another study, as well as in this report, 16:1 predominated in the E. coli media (Mayer and Shanklin 2007; Figs. 5, 6). Therefore, the saturation level of substrates utilized by ALT1–4 in E. coli may be different from their true in planta substrates. This is especially evident when considering that unsaturated fatty acids of chains lengths shorter than C18, while present in E. coli, are not significant metabolites in plant cells. However, the deduced substrate specificities with regards to chain length and oxidation state are expected to be roughly in accordance with in planta substrates.
Despite the very high similarities between the ALTs in their protein sequences, the products they generated in E. coli were substantially different and this likely reflects differing specificities in planta. Fatty acids were the major products of ALT1 (C12, C14, and C16 chain lengths) as well as ALT4 (C6 and C8 chain lengths). Prior to this study, the shortest chain fatty acid reported to be a major product of a plant acyl-TE was 8:0 generated by Cuphea spp. FATB variants (Tjellström et al. 2013). The 6:0 fatty acid generated by ALT4 is now the shortest known major fatty acid product of a plant fatty acyl-TE. The major products of ALT2 were 8:0 and 10:0 β-ketofatty acids. We chemically converted the inherently unstable β-ketofatty acids produced in E. coli to methylketones for detection and quantification by gas chromatography. In the plant, the β-ketofatty acids are expected to be further metabolized and methylketones are also the likely end product. In wild tomato, β-ketofatty acids produced by ShMKS2, which is highly related to the ALTs, are converted to methylketones by a β-ketofatty acid decarboxylase (ShMKS1). ShMKS1 is an atypical member of the α/β–hydrolase superfamily (Auldridge et al. 2012). Arabidopsis contains many genes encoding predicted α/β–hydrolases, but it is currently unknown whether any of these can act as β-ketofatty acid decarboxylases. Sequence similarity to ShMKS1 identifies several of the 21 METHYL ESTERASE (MES) gene family members as potential orthologs in Arabidopsis, although no single candidate is obvious by sequence inspection or by co-regulation analysis. ALT3 appears to have broader substrate specificity, overlapping with the other ALT family members. All of the products generated by ALT1, ALT2, and ALT4 were produced at some level by ALT3. The apparent broad specificity of ALT3 may indicate its involvement in more than one biological role and it may have functional overlap with the other ALTs.
Lauroyl-ACP TE activity was demonstrated in vitro for ALT1, 3, and 4, but not for ALT2. In our E. coli expression experiment, 12:0 fatty acid was the major product of the ALT1 expressing strain, with levels of this product elevated approximately 15-fold compared to the vector only control. In contrast, 12:0 fatty acid was a minor product of ALT3 and 4, with levels only 1.8 and 2.3 times that of the vector only control. However, in our in vitro assay, ALT3 and ALT4 displayed significantly higher 12:0-ACP TE activity than did ALT1. This discrepancy may be explained by difficulties in obtaining high quality enzyme preparations. We obtained a lower yield and purity for ALT1 than we did for ALT3 or ALT4, and our ALT1 preparations may contain a larger number of misfolded and/or inactive enzyme molecules. The levels of 12:0 fatty acid produced by the ALT2 strain were barely above the vector only control levels, which is consistent with the lack of 12:0-ACP TE activity in vitro. It should be noted that of the four ALT proteins, ALT2 was the only one to not produce any fully reduced fatty acids as a major product when expressed in E. coli. Future studies examining the preferred substrates of the ALT proteins need to be conducted by assaying in vitro different substrates and comparing relative enzyme kinetic activities.
ALT proteins likely form homodimers or heterodimers, as this is common for Hotdog fold enzymes (Pidugu et al. 2009). Each ALT produced novel lipid products when expressed alone in E. coli, indicating that they can function as homodimers. However, heterodimeric complexes have been reported for Hotdog fold proteins (Sacco et al. 2007), and this could be the case for ALTs in planta. The ubiquitous expression of ALT3 would allow for the encoded protein to form heterodimers with ALT1, 2, or 4.
The ALT family spans broadly across plants, but functions might not be conserved
Homologues of the Arabidopsis ALTs are found in wide ranging plant taxa. In a phylogenetic tree, the ALT proteins cluster mainly with proteins from the same or related species. This is in contrast to FATA- and FATB-type thioesterases, where proteins cluster according to functional FATA-type and FATB-type clades, even across vast plant lineages (Mayer and Shanklin 2007, Supplementary Fig. S1). This suggests that there is an ancient ALT-type gene ancestor, but that gene duplication events leading to paralogues occurred relatively recently. The ubiquitous expression and the relatively broad substrate specificity of ALT3 suggests that it is most related in activity and function to an ancient, multifunctional enzyme. ALT1, ALT2 and ALT4, which are more restricted in both expression patterns and substrate preferences, may have evolved though sub-functionalization to have more specialized activities and biological roles. Atypical FATB enzymes with specificity for shorter chain lengths substrate (e.g. Cuphea hookeriana FATB2 and Umbellularia californica FATB, which produce 8:0–10:0 and 10:0–12:0, respectively), are evolutionarily distinct from ALT1 and ALT4, despite having similar substrate preferences, indicating that this specialization arose independently (Supplementary Fig. S1).
Possible biological roles for ALT1–4
The gene expression pattern of ALT1 is suggestive of a role in cuticular wax synthesis. Expression in aerial tissues, in particular epidermal cells, is a characteristic of cuticle biosynthetic genes. This is consistent with public microarray data that shows expression of ALT1 correlated with expression of genes involved in cuticular wax biosynthesis, a process that occurs late in cuticle development. The biochemistry of wax biosynthesis is well characterized (Bernard and Joubès 2012). Wax precursors are delivered as very-long-chain acyl-CoAs to the ER where chemical modification occurs. It thus seems unclear how a plastid localized acyl-ACP TE that produces medium chain fatty acids would be involved in this process. It is possible that ALT1 is involved in generating non-wax metabolites that accumulate in the cuticle. The high volatility of these compounds would cause them to be missed in typical cuticular wax extraction procedures that involve dipping of plant tissues in chloroform or hexane followed by evaporation under nitrogen gas.
The gene expression pattern of ALT2 suggests a role in suberin biosynthesis. In addition to strong co-regulation coefficients with known suberin genes, ALT2 is expressed at known sites of suberin deposition: endodermal and peridermal cell layers of roots as well as seeds. However, the plastid localization and production of short chain β-ketofatty acids in E. coli are inconsistent with a role in suberin synthesis, which uses long-chain fatty acids as starting material and occurs in the ER (Beisson et al. 2012). A commonality with the sites of ALT2 expression is that these are regions that are fortified against microbial pathogen and insect attack. ALT2 may generate β-ketofatty acids at these sites that are converted into methylketones for defense against pests. It is interesting to note that 5:0 methylketone has been detected in volatile emissions from A. thaliana after herbivore damage (van Poecke et al. 2001), and ALT2 is a good candidate enzyme for producing this compound. It would be expected that a decarboxylase, likely a homolog of ShMKS1 (Auldridge et al. 2012), would catalyze the conversion of β-ketofatty acid to methylketone in planta.
As postulated earlier, ALT3 function may be at least partially redundant to ALT1, ALT2 and ALT4 in tissues where gene expression overlap, but it likely also has a unique role. The main product of ALT3 in our E. coli experiment was uniquely 14:1 β-ketofatty acid (chemically converted to methylketone in our experimental procedure). It is possible that this is converted to a C13 methylketone in planta, like that produced in wild tomato, with the naturally produced β-ketofatty acid likely to be saturated. Metabolomic profiles of Arabidopsis indicate the presence of a 15:0 methyl ketone (Bais et al. 2010) and ALT3 is a candidate for the production of this compound as it produced 16:0 β-ketofatty acids in E. coli. The presence of additional methylketones in Arabidopsis could easily have been overlooked as they are highly volatile, which may cause them to be lost during evaporation of solvents, a common step in extraction protocols. The shorter chain length methylketones would also elute too early to be detected by GC analysis using routine methods. ALT3 showed significant activity towards 16:1 fatty acyl-ACP in our E. coli experiments. If, like FATB, the degree of saturation of the preferred substrate is altered in this heterologous system, and ALT3 acts on 16:0 fatty acyl-ACPs in planta, the two proteins may be partially redundant. In a fatb mutant with less than 1 % of wild-type transcript levels, saturated fatty acids and their derivatives are reduced by only 20–50 % depending on the tissue type and lipid species analyzed (Bonaventure et al. 2003). Saturated fatty acids are reduced further, but not eliminated, in a fatb act1 double mutant, which additionally lacks plastid glycerol-3-phosphate:acyl-ACP acyltransferase activity (Bonaventure et al. 2003). These authors postulated that mitochondrial fatty acid synthesis may compensate for the low 16:0 acyl-ACP TE activity in plastids or that the low-level activity of FATA for saturated fatty acyl-ACPs provides some saturated fatty acids. We now propose that ALT3 generates a proportion of the saturated fatty acids that exit the plastid.
ALT4 is expressed in anthers, and generates 6:0 and 8:0 saturated fatty acids when expressed in E. coli. ALT4 may play a role in development of floral organs by generating short chain fatty acids to modulate ethylene sensing and production in anthers. Short chain saturated fatty acids are known to be produced by floral organs of certain ornamental flowers, and play roles in ethylene perception, for example, during the transition from pre-fruit maturation to fruit maturation and senescence (Whitehead and Halevy 1989; Whitehead and Vasiljevic 1993; Halevy et al. 1996). Ethylene has been shown to promote anther dehiscence in tobacco, although jasmonic acid and not ethylene is thought to promote anther dehiscence in Arabidopsis (Rieu et al. 2003). Another possibility is that ALT4 may be involved in production of odor compounds to attract insect pollinators or insect bodyguards. Although Arabidopsis generally self-pollinates, insect mediated cross-pollination is observed in wild populations (Tan et al. 2005), and fatty acids and their derivatives are often part of floral scent mixtures (Knudsen et al. 1993).
Conclusion
Acyl-lipid thioesterase1 (ALT1), ALT2, ALT3, and ALT4 represent a novel family of plastid-localized acyl-ACP thioesterases in A. thaliana. These proteins produce different lipid products despite their high amino acid sequence similarities. They may be involved in generating specialized lipid compounds previously unreported in Arabidopsis. There are currently no publically available T-DNA insertion lines for ALT1–4; therefore mutant analysis will require gene silencing strategies. Predicted ALT-type proteins are found in a wide variety of plant species. Considering the enzymatic diversity within one plant species, the variety of potential activities of other ALT-type enzymes is immense.
References
Aarts MG, Keijzer CJ, Stiekema WJ, Pereira A (1995) Molecular characterization of the CER1 gene of Arabidopsis involved in epicuticular wax biosynthesis and pollen fertility. Plant Cell 7(12):2115–2127
Aharoni A, Dixit S, Jetter R, Thoenes E, van Arkel G, Pereira A (2004) The SHINE clade of AP2 domain transcription factors activates wax biosynthesis, alters cuticle properties, and confers drought tolerance when overexpressed in Arabidopsis. Plant Cell 16(9):2463–2480
Auldridge ME, Guo Y, Austin MB, Ramsey J, Fridman E, Pichersky E, Noel JP (2012) Emergent decarboxylase activity and attenuation of α/β-hydrolase activity during the evolution of methylketone biosynthesis in tomato. Plant Cell 24(4):1596–1607
Bais P, Moon SM, He K, Leitao R, Dreher K, Walk T, Sucaet Y, Barkan L, Wohlgemuth G, Roth MR, Wurtele ES, Dixon P, Fiehn O, Lange BM, Shulaev V, Sumner LW, Welti R, Nikolau BJ, Rhee SY, Dickerson JA (2010) PlantMetabolomics.org: a web portal for plant metabolomics experiments. Plant Physiol 152(4):1807–1816
Beisson F, Li Y, Bonaventure G, Pollard M, Ohlrogge JB (2007) The acyltransferase GPAT5 is required for the synthesis of suberin in seed coat and root of Arabidopsis. Plant Cell 19(1):351–368
Beisson F, Li-Beisson Y, Pollard M (2012) Solving the puzzles of cutin and suberin polymer biosynthesis. Curr Opin Plant Biol 15(3):329–337
Ben-Israel I, Yu G, Austin MB, Bhuiyan N, Auldridge M, Nguyen T, Schauvinhold I, Noel JP, Pichersky E, Fridman E (2009) Multiple biochemical and morphological factors underlie the production of methylketones in tomato trichomes. Plant Physiol 151(4):1952–1964
Benning C (2009) Mechanisms of lipid transport involved in organelle biogenesis in plant cells. Annu Rev Cell Dev Biol 25:71–91
Benning MM, Wesenberg G, Liu R, Taylor KL, Dunaway-Mariano D, Holden HM (1998) The three-dimensional structure of 4-hydroxybenzoyl-CoA thioesterase from Pseudomonas sp. Strain CBS-3. J Biol Chem 273(50):33572–33579
Bernard A, Joubès J (2012) Arabidopsis cuticular waxes: advances in synthesis, export and regulation. Prog Lipid Res 52(1):110–129
Bonaventure G, Salas JJ, Pollard MR, Ohlrogge JB (2003) Disruption of the FATB gene in Arabidopsis demonstrates an essential role of saturated fatty acids in plant growth. Plant Cell 15(4):1020–1033
Cantu DC, Chen Y, Reilly PJ (2010) Thioesterases: a new perspective based on their primary and tertiary structures. Protein Sci 19(7):1281–1295
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16(6):735–743
Compagnon V, Diehl P, Benveniste I, Meyer D, Schaller H, Schreiber L, Franke R, Pinot F (2009) CYP86B1 is required for very long chain omega-hydroxyacid and alpha, omega-dicarboxylic acid synthesis in root and seed suberin polyester. Plant Physiol 150(4):1831–1843
Dean GH, Zheng H, Tewari J, Huang J, Young DS, Hwang YT, Western TL, Carpita NC, McCann MC, Mansfield SD, Haughn GW (2007) The Arabidopsis MUM2 gene encodes a beta-galactosidase required for the production of seed coat mucilage with correct hydration properties. Plant Cell 19(12):4007–4021
Dillon SC, Bateman A (2004) The Hotdog fold: wrapping up a superfamily of thioesterases and dehydratases. BMC Bioinformatics 5:109
Domergue F, Vishwanath SJ, Joubès J, Ono J, Lee JA, Bourdon M, Alhattab R, Lowe C, Pascal S, Lessire R, Rowland O (2010) Three Arabidopsis fatty acyl-coenzyme A reductases, FAR1, FAR4, and FAR5, generate primary fatty alcohols associated with suberin deposition. Plant Physiol 153(4):1539–1554
Dörmann P, Voelker TA, Ohlrogge JB (1995) Cloning and expression in Escherichia coli of a novel thioesterase from Arabidopsis thaliana specific for long-chain acyl–acyl carrier proteins. Arch Biochem Biophys 316(1):612–618
Emanuelsson O, Nielsen H, Brunak S, von Heijne G (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300:1005–1016
Fellermeier M, Eisenreich W, Bacher A, Zenk MH (2001) Biosynthesis of cannabinoids. Incorporation experiments with (13)C-labeled glucoses. Eur J Biochem 268(6):1596–1604
Fridman E, Wang J, Iijima Y, Froehlich JE, Gang DR, Ohlrogge J, Pichersky E (2005) Metabolic, genomic, and biochemical analyses of glandular trichomes from the wild tomato species Lycopersicon hirsutum identify a key enzyme in the biosynthesis of methylketones. Plant Cell 17(4):1252–1267
Goh EB, Baidoo EE, Keasling JD, Beller HR (2012) Engineering of bacterial methyl ketone synthesis for biofuels. Appl Environ Microbiol 78(1):70–80
Greer S, Wen M, Bird D, Wu X, Samuels L, Kunst L, Jetter R (2007) The cytochrome P450 enzyme CYP96A15 is the midchain alkane hydroxylase responsible for formation of secondary alcohols and ketones in stem cuticular wax of Arabidopsis. Plant Physiol 145(3):653–656
Halevy AH, Porat R, Spiegelslein H, Borochov A, Botha L, Whitehead CS (1996) Short-chain saturated fatty acids in the regulation of pollination induced ethylene sensitivity of Phalaenopsis flowers. Physiol Plant 97(3):469–474
Höfer R, Briesen I, Beck M, Pinot F, Schreiber L, Franke R (2008) The Arabidopsis cytochrome P450 CYP86A1 encodes a fatty acid omega-hydroxylase involved in suberin monomer biosynthesis. J Exp Bot 59(9):2347–2360
Hooker TS, Millar AA, Kunst L (2002) Significance of the expression of the CER6 condensing enzyme for cuticular wax production in Arabidopsis. Plant Physiol 129:1568–1580
Hunt MC, Siponen MI, Alexson SE (2012) The emerging role of acyl-CoA thioesterases and acyltransferases in regulating peroxisomal lipid metabolism. Biochim Biophys Acta 1822(9):1397–1410
Jones A, Davies HM, Voelker TA (1995) Palmitoyl-acyl carrier protein (ACP) thioesterase and the evolutionary origin of plant acyl-ACP thioesterases. Plant Cell 7(3):359–371
Knudsen JT, Tollsten L, Bergstrom LG (1993) Floral scents: a checklist of volatile compounds isolated by headspace techniques. Phytochemistry 33(2):253–280
Kurata T, Kawabata-Awai C, Sakuradani E, Shimizu S, Okada K, Wada T (2003) The YORE–YORE gene regulates multiple aspects of epidermal cell differentiation in Arabidopsis. Plant J 36(1):55–66
Lenfant N, Hotelier T, Velluet E, Bourne Y, Marchot P, Chatonnet A (2013) ESTHER, the database of the α/β-hydrolase fold superfamily of proteins: tools to explore diversity of functions. Nucleic Acids Res 41(Database issue):D423–9
Li F, Wu X, Lam P, Bird D, Zheng H, Samuels L, Jetter R, Kunst L (2008) Identification of the wax ester synthase/acyl-coenzyme A: diacylglycerol acyltransferase WSD1 required for stem wax ester biosynthesis in Arabidopsis. Plant Physiol 148(1):97–107
Li-Beisson Y, Shorrosh B, Beisson F, Andersson M, Arondel V, Bates P, Baud S, Bird D, DeBono A, Durrett T et al (2013) Acyl-lipid metabolism. Arabidopsis Book 11:e0161
Lü S, Song T, Kosma DK, Parsons EP, Rowland O, Jenks MA (2009) Arabidopsis CER8 encodes LONG-CHAIN ACYL-COA SYNTHETASE 1 (LACS1) that has overlapping functions with LACS2 in plant wax and cutin synthesis. Plant J 59(4):553–564
Marks MD, Tian L, Wenger JP, Omburo SN, Soto-Fuentes W, He J, Gang DR, Weiblen GD, Dixon RA (2009) Identification of candidate genes affecting delta9-tetrahydrocannabinol biosynthesis in Cannabis sativa. J Exp Bot 60(13):3715–3726
Mayer KM, Shanklin J (2007) Identification of amino acid residues involved in substrate specificity of plant acyl-ACP thioesterases using a bioinformatics-guided approach. BMC Plant Biol 7:1
Molina I, Li-Beisson Y, Beisson F, Ohlrogge JB, Pollard M (2009) Identification of an Arabidopsis feruloyl-coenzyme A transferase required for suberin synthesis. Plant Physiol 151(3):1317–1328
Moreno-Pérez AJ, Venegas-Calerón M, Vaistij FE, Salas JJ, Larson TR, Garcés R, Graham IA, Martínez-Force E (2012) Reduced expression of FatA thioesterases in Arabidopsis affects the oil content and fatty acid composition of the seeds. Planta 235(3):629–639
Nardini M, Dijkstra BW (1999) Alpha/beta hydrolase fold enzymes: the family keeps growing. Curr Opin Struct Biol 9(6):732–737
Nelson BK, Cai X, Nebenführ A (2007) A multicolored set of in vivo organelle markers for co-localization studies in Arabidopsis and other plants. Plant J 51(6):1126–1136
Nielsen H, Engelbrecht J, Brunak S, von Heijne G (1997) Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng 10(1):1–6
Overath P, Pauli G, Schairer HU (1969) Fatty acid degradation in Escherichia coli. An inducible acyl-CoA synthetase, the mapping of old-mutations, and the isolation of regulatory mutants. Eur J Biochem 7(4):559–574
Pidugu LS, Maity K, Ramaswamy K, Surolia N, Suguna K (2009) Analysis of proteins with the ‘hot dog’ fold: prediction of function and identification of catalytic residues of hypothetical proteins. BMC Struct Biol 9:37
Rieu I, Wolters-Arts M, Derksen J, Mariani C, Weterings K (2003) Ethylene regulates the timing of anther dehiscence in tobacco. Planta 217(1):131–137
Rowland O, Zheng H, Hepworth SR, Lam P, Jetter R, Kunst L (2006) CER4 encodes an alcohol-forming fatty acyl-coenzyme A reductase involved in cuticular wax production in Arabidopsis. Plant Physiol 142(3):866–877
Sacco E, Covarrubias AS, O’Hare HM, Carroll P, Eynard N, Jones TA, Parish T, Daffé M, Bäckbro K, Quémard A (2007) The missing piece of the type II fatty acid synthase system from Mycobacterium tuberculosis. Proc Natl Acad Sci USA 104(37):14628–14633
Samach A, Kohalmi SE, Motte P, Datla R, Haughn GW (1997) Divergence of function and regulation of class B floral organ identity genes. Plant Cell 9(4):559–570
Sparkes IA, Runions J, Kearns A, Hawes C (2006) Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat Protoc 1(4):2019–2025
Tan YY, Xu HH, Tao WJ, Hoffmann MH, Wang XF, Lu YT (2005) Transgenic GFP as a molecular marker for approaches to quantify pollination mechanism and gene flow in Arabidopsis thaliana. Plant Biol (Stuttg) 7(4):405–410
Tilton GB, Shockey JM, Browse J (2004) Biochemical and molecular characterization of ACH2, an acyl-CoA thioesterase from Arabidopsis thaliana. J Biol Chem 279(9):7487–7494
Tjellström H, Strawsine M, Silva J, Cahoon EB, Ohlrogge JB (2013) Disruption of plastid acyl:acyl carrier protein synthetases increases medium chain fatty acid accumulation in seeds of transgenic Arabidopsis. FEBS Lett 587(7):936–942
Toufighi K, Brady SM, Austin R, Ly E, Provart NJ (2005) The Botany Array Resource: e-Northerns, Expression Angling, and promoter analyses. Plant J 43(1):153–163
Van Poecke RM, Posthumus MA, Dicke M (2001) Herbivore-induced volatile production by Arabidopsis thaliana leads to attraction of the parasitoid Cotesia rubecula: chemical, behavioral, and gene-expression analysis. J Chem Ecol 27(10):1911–1928
Voelker TA, Worrell AC, Anderson L, Bleibaum J, Fan C, Hawkins DJ, Radke SE, Davies HM (1992) Fatty acid biosynthesis redirected to medium chains in transgenic oilseed plants. Science 257(5066):72–74
Whitehead CS, Halevy AH (1989) Ethylene sensitivity: the role of short-chain saturated fatty acids in pollination-induced senescence of Petunia hybrida flowers. Plant Growth Regul 8(1):41–54
Whitehead CS, Vasiljevic D (1993) Role of short-chain saturated fatty acids in the control of ethylene sensitivity in senescing carnation flowers. Physiol Plant 88(20):243–250
Yu G, Nguyen TT, Guo Y, Schauvinhold I, Auldridge ME, Bhuiyan N, Ben-Israel I, Iijima Y, Fridman E, Noel JP, Pichersky E (2010) Enzymatic functions of wild tomato METHYLKETONE SYNTHASES 1 and 2. Plant Physiol 154(1):67–77
Acknowledgments
We thank Denise Chabot (Eastern Cereal and Oilseed Research Centre, Agriculture and Agri-Food Canada) for assistance with the confocal microscope. We thank Carlos Canez (Carleton University) for assistance with propagation and imaging of promoter:: GUS transgenic plants. We thank Dr. Shelley Hepworth (Carleton University) for critical reading of the manuscript. This work was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada to Owen Rowland.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Pulsifer, I.P., Lowe, C., Narayaran, S.A. et al. Acyl-lipid thioesterase1–4 from Arabidopsis thaliana form a novel family of fatty acyl–acyl carrier protein thioesterases with divergent expression patterns and substrate specificities. Plant Mol Biol 84, 549–563 (2014). https://doi.org/10.1007/s11103-013-0151-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-013-0151-z