
Abstract
The twin-arginine protein translocation (Tat) system has been characterized in bacteria, archaea and the chloroplast thylakoidal membrane. This system is distinct from other protein transport systems with respect to two key features. Firstly, it accepts cargo proteins with an N-terminal signal peptide that carries the canonical twin-arginine motif, which is essential for transport. Second, the Tat system only accepts and translocates fully folded cargo proteins across the respective membrane. Here, we review the core essential features of folded protein transport via the bacterial Tat system, using the three-component TatABC system of Escherichia coli and the two-component TatAC systems of Bacillus subtilis as the main examples. In particular, we address features of twin-arginine signal peptides, the essential Tat components and how they assemble into different complexes, mechanistic features and energetics of Tat-dependent protein translocation, cytoplasmic chaperoning of Tat cargo proteins, and the remarkable proofreading capabilities of the Tat system. In doing so, we present the current state of our understanding of Tat-dependent protein translocation across biological membranes, which may serve as a lead for future investigations.
Similar content being viewed by others
1 Introduction
To function correctly and efficiently, every cell needs to be highly organised, tightly regulated and compartmentalised. Proteins are essential macromolecules synthesised by ribosomes in the cytoplasm that often require localisation to a particular subcellular compartment before they can carry out their respective functions. Their proper formation, targeting and activity are imperative to the survival of the cell. This requirement for correct localisation particularly applies to proteins that take part in the acquisition of nutrients, energy transduction, cell-to-cell communication and cellular locomotion. On average, 20–30% of proteins synthesised in the bacterial cytoplasm are destined for extra-cytoplasmic locations [1]. They therefore have to pass a cell membrane composed of a tightly sealed lipid bilayer intent on keeping the cell structurally sound and impenetrable. Therefore, specialised transport systems have evolved within the cell membrane to allow proteins to cross this barrier. Each system made up of critical components is as specialised as the protein cargo it will transport. However common features tie protein transport systems together, which guarantee cell regulation and safety. These include a gated pore, an energy requirement to drive cargo proteins through the membrane, and the use of signal peptides that direct the cargo protein to the correct translocase and the correct location.
Two major transport systems exist for protein translocation across the bacterial cytoplasmic membrane, namely the general secretory (Sec) pathway and the twin-arginine translocation (Tat) pathway (Fig. 1). The Sec pathway facilitates export of the majority of bacterial proteins, whereas the Tat pathway is quite restricted. For instance, it transports ~ 30 proteins in Escherichia coli and only four in Bacillus subtilis [2]. Further, each protein is fully folded in the cytoplasm prior to export via Tat, whereas Sec can only export unfolded proteins.

The Sec- and Tat-dependent protein transport pathways. The Sec pathway is the dominant pathway for protein export from the bacterial cytoplasm. It accepts and translocates cargo proteins across the plasma membrane in a loosely folded or unfolded state, here exemplified with the precursor of the outer membrane protein A of E. coli (OmpA). Targeting and folding control of the cargo protein is supported by cytoplasmic targeting factors, such as SecB. The Sec machinery itself is composed of the SecYEG channel and the translocation ATPase SecA, which converts chemical energy in the form of ATP into a driving force that pushes the cargo protein through the membrane. Additionally, translocation may be powered by the transmembrane proton gradient. At the trans-side of the membrane, the translocated protein folds into its active and protease-resistant final conformation. In contrast to the Sec pathway, the Tat pathway transports fully folded cofactor-containing proteins across the membrane, here exemplified with the precursor of the Tat cargo TorA. Cofactor insertion and folding may be aided by Redox Enzyme Maturation Proteins (REMPS), such as TorD in the case of TorA. The Tat translocase may consist of the three components TatA, TatB and TatC (E. coli), or of TatA and TatC components only (B. subtilis). Protein transport via Tat is powered by the transmembrane proton-motive force
2 Protein Targeting Via the Twin-Arginine Signal Peptide
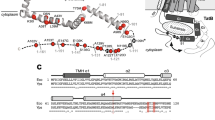
To ensure proteins are appropriately directed into the Sec or Tat pathways and to initiate the translocation process, specific signal peptides are present on the N-terminus of each protein. On the trans side of the membrane the signal peptide is cleaved by a signal peptidase to liberate just the mature protein [3,4,5,6,7]. The amino acid sequences of signal peptides differ substantially, but they are all composed of a positively charged N-terminal N-domain, a hydrophobic H-domain and a C-terminal C-domain with an Ala-x-Ala signal peptidase cleavage site [3, 8] (Fig. 2). Further, the N-regions of Tat signal peptides contain the canonical twin-arginine motif S-R-R-x-F-L-K (where x is a polar amino acid) [9]. The importance of additional conserved amino acids in the Tat-motif depends on the cargo protein and varies in different bacteria [10]. However, RR-residues are close to invariant and key to efficient protein export. In particular, the charge-neutral substitution of RR to KK blocks Tat export completely [11]. Yet, a single Arg to Lys mutation only slows down the rate of translocation in most bacteria [12]. In chloroplast thylakoids where the Tat pathway also exists, an RR to KR substitution is tolerated, while a RR to RK substitution precludes transport [12,13,14]. A single substitution of Arg to Glu has been reported as tolerated too [15]. Of note, the TtrB subunit of the tetrathionate reductase in Enterobacteriaceae is the only known native Tat cargo to have a KR-motif [16]. Aside from the RR-motif, other residues within the larger twin-arginine signal peptide are also important. In particular, the Phe residue is present in 80% of Tat-motifs, and substitutions showed a highly hydrophobic residue is essential at this position [11].

Sec- and Tat-specific signal peptides. N-terminal signal peptides direct proteins to the Sec and Tat translocases in the membrane. They have a conserved tripartite structure, consisting of a positively charged N-region (indicated by ‘white residues’ in one-letter code), a hydrophobic H-region (red) and a C-region (green) that contains the Ala-X-Ala recognition site for signal peptidase. Cleavage by signal peptidase, C-terminally from the Ala-X-Ala sequence, liberates the mature protein (pink) from the membrane. Twin-arginine signal peptides, as exemplified by the TorA signal peptide, contain the canonical twin-arginine motif at the interface of the N- and C-regions. Their H-region is longer and less hydrophobic than that of Sec-type signal peptides, and N-terminally of the C-region there are often positively charged residues that serve in Sec-avoidance. Notably, Sec-type signal peptides, here exemplified by the OmpA signal peptide, are usually much shorter than twin-arginine signal peptides
Tat signal peptides comprise about 30 residues in most organisms. Hence they are longer than Sec signal peptides, which comprise about 17 to 24 residues [17]. Tat signal peptides are also overall less hydrophobic than Sec signal peptides, which serves to avoid protein targeting to the Sec pathway [18]. Additionally, the C-domain of Tat signal peptides may include basic residues N-terminally of the A-x-A motif, which contribute to Sec avoidance (Fig. 2) [19, 20].
3 The Twin-Arginine Translocation Pathway
In the early 1990’s, an alternative translocase was discovered in the thylakoid membrane of chloroplasts, which worked in parallel to the Sec pathway [21]. Initially this pathway was named the ΔpH-dependent pathway due to its unusual sole requirement of a transmembrane proton gradient for translocation [22]. Three membrane proteins were soon identified in thylakoids as essential for translocation of fully folded proteins via the ΔpH-dependent pathway [23], namely Tha4 [24], Hcf106 [25] and cpTatC [26]. Subsequently, homologous proteins were identified in some bacteria, archaea and even mitochondria [27, 28]. In E. coli, the homologues of Tha4, Hcf106 and cpTatC were also shown to be required for export of proteins with twin-arginine signal peptides and, therefore, they were respectively named TatA, TatB and TatC [9, 29,30,31].
Combined studies on the thylakoidal and bacterial Tat pathways showed that their function is to transport a subset of complex fully folded proteins that require cofactor insertion or immediate oligomerisation [8, 32]. Today, Tat-translocated proteins have been shown to participate in many processes including energy metabolism, cell division, cell envelope biogenesis, quorum sensing, motility, symbiosis and pathogenesis [33,34,35,36]. Tat can even export complex heterologous proteins that are Sec-incompatible, like the tightly folded dihydrofolate reductase with bound methotrexate [37], the green fluorescent protein (GFP) [38], and several bio-pharmaceutically relevant human proteins [39]. Another intriguing attribute of the Tat pathway is that it can detect unfolded or mutated proteins, and reject them for export [40, 41].
Based on the number of Tat components involved in protein translocation, essentially two types of ‘translocases’ can be distinguished. The prototype Tat translocase that is active in thylakoids and E. coli, consists of the afore-mentioned TatABC components. Further, the minimal Tat translocases, as typified in Bacillus species consist of TatA and TatC components only. The types of translocases will be discussed in the following paragraphs.
4 The E. coli Tat Translocase
The E. coli tatABCD operon encodes the core components of this bacterium’s Tat system (Table 1). All four genes are constitutively expressed, but the expression level of tatA exceeds that of tatB 25-fold that of tatC 50-fold [42]. This difference is mirrored in the final component make-up of the Tat translocase in the plasma membrane. The tatE gene is constitutively expressed from another chromosomal locus. The tatB and tatE genes are thought to originate from gene duplications of tatA [28, 43]. Although ΔtatABCDE strains are viable, the mutants show various defects including impaired septation, decreased motility and an increased sensitivity to detergent [44].
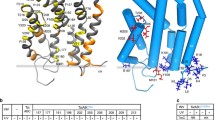
TatA (9.6 kDa) is the most abundant component of the Tat complex, most likely responsible for forming the translocase channel [45]. E. coli has a core TatA protein, but it also involves the TatA-like proteins, TatB and TatE [28]. Of note, TatE can substitute TatA [43]. TatA, TatB and TatE are similar in structure with a short N-terminal domain that is exposed to the periplasm [46], a single transmembrane helix, an amphipathic helix in the cytoplasm [47], and an unstructured cytoplasmic C-domain [48] (Fig. 3). Surprisingly, not many mutations in TatA block export, but there are a few instances. In particular, Gly33 in the “hinge region” is critical for TatA function [49], and the transmembrane helix and various residues in the amphipathic helix are also important [50, 51].

Membrane topology and structures of the TatA, TatB and TatC proteins. The Tat translocase of E. coli consists of three components, namely TatA, TatB and TatC. TatB and TatC form a receptor complex for cargo proteins, whereas TatA is the main facilitator for protein translocation across the membrane. TatB is missing from the two-component Tat translocases as encountered in B. subtilis. The upper half of the Figure shows a traditional representation of the membrane topology of TatA/E, TatB and TatC based on molecular biological analyses. The lower half of the Figure shows ribbon presentations of the structures of TatA, TatB and TatC as adopted from the RCSB Protein Data Bank (http://www.rcsb.org/structure/). These structures have the following PDB accession codes: TatA—2LZR (solution NMR structure [48]); TatB—2MI2 (solution NMR structure [54]); and TatC—4HTS (crystal structure [63])
TatE (7 kDa) is a much smaller than TatA [9]. Given the smaller size and ~ 100-fold lower abundance than TatA, it was initially believed TatE has no real function in the Tat translocon [42]. More recently however, it was shown TatE could substitute TatA [43], and that it is recruited to the Tat translocase [52]. Importantly, TatE was shown to interact with the Tat signal peptide and to even partially prevent premature cleavage of the TorA signal peptide [53].
The role of TatB (18.4 kDa) is to bind the Tat signal peptide and, thereafter, the mature protein. Despite only sharing 20% sequence identity to TatA and being nearly double TatA’s size, TatB is predicted to have a very similar structure and topology (Fig. 3) [50]. Specifically, TatB has a slightly longer amphipathic helix and a longer unstructured C-terminal region [54, 55]. Mutations in TatB’s hinge region and amphipathic helix cause translocation defects [56]. Of note, particular amino acid substitutions in TatA’s N-terminus allow replacement of TatB by TatA [57] [58], supporting the notion that TatB originated from TatA and subsequently specialized [5, 59].
TatC is the largest (28.9 kDa) and best-conserved protein in the Tat complex that aids cargo binding [60, 61]. The structure of TatC is very different to other Tat components as it has 6 transmembrane helices and an N-in C-in topology (Fig. 3) [62]. The crystallisation of TatC from Aquifex aeolicus, which shares 40% sequence identity to E. coli Tat C, revealed the relative positions of the transmembrane domains [63]. Together, they take the shape of a baseball glove or cupped hand with very restricted structural flexibility [64]. Notably, a conserved Glu residue (Glu170 in E. coli) is positioned close to the signal peptide binding pocket in the plane of the membrane and potentially perturbs the bilayer structure [12, 64, 65]. Additional residues needed for TatC function reside in the cytoplasmic N-region and the first cytoplasmic loop [61, 66].
5 The B. subtilis Tat Translocase
The Tat translocase can minimally function with just TatA and TatC [5, 67,68,69]. Interestingly, the Gram-positive bacterium B. subtilis has two minimal Tat translocases encoded by the tatAdCd and tatAyCy operons, which work in parallel and with different cargo specificities (Table 1) [5]. TatAdCd has only one known cargo protein, PhoD, which is co-expressed with the translocase under phosphate-deprived conditions [68, 70]. TatAyCy is constitutively expressed, along with its cargo proteins EfeB (YwnN), QcrA and YkuE [5, 71,72,73,74]. The third TatA gene of B. subtilis, tatAc, is constitutively expressed from another locus, and was shown to serve a non-essential function in protein translocation via the TatAyCy [5, 75].
B. subtilis TatAd and TatAy are bifunctional, meaning that they act at the same time as E. coli TatA and TatB. Interestingly, B. subtilis TatAd can replace TatA and TatB in E. coli [76], whereas TatAc expressed in E. coli can functionally replace TatA and TatE and form active translocases with TatCd and TatCy [77, 78]. This suggests that, despite species-specific features, the translocation mechanism employed by Tat is conserved across species [76, 79].
Structural studies on B. subtilis TatAd (7.4 kDa) have confirmed its ‘L-shape’ arrangement in the membrane [80,81,82]. By itself, TatAd oligomerizes to complexes of ~ 270 kDa and, together with TatCd (28 kDa), TatAd forms complexes of ~ 230 kDa in which TatAd is stabilized by TatCd [83,84,85]. Although the structure of TatAd resembles that of E. coli TatA, the effects of particular amino acid substitutions differ for the two proteins [47, 86]. Notably, mutagenesis of the TatAd N-terminus blocks protein translocation in E. coli tatB mutant cells, indicating that the N-terminal residues of TatAd are needed for TatB substitution [83].
Like TatAd, TatAy (6 kDa) has a structure similar to that of E. coli TatA [83, 86]. In particular, the conserved Pro2 residue in the N-terminus of TatAy and its hinge region are required for protein export [75, 86]. Complexes of TatAy alone and TatAyCy have a molecular mass of ~ 200 kDa [87]. Intriguingly, a P2A mutation leads to the formation of large fibrils composed of TatAy and TatCy, suggesting that Pro2 serves a role in the termination of complex assembly [88].
TatCd and TatCy (28/28.9 kDa) resemble E. coli TatC, having six transmembrane helices [87, 85]. Further, the N-terminus, the first cytoplasmic loop and the C-terminal tail of TatCd and TatCy are important for protein export, but the relevance of different conserved residues depends on the cargo [89, 90].
TatAc (6.7 kDa) of B. subtilis shares significant sequence similarity with E. coli TatE, and it can actually form active Tat complexes with TatA and TatB, or with TatCd and TatCy when expressed in E. coli (Table 1) [75,76,77,78, 87]. Nevertheless, TatAc cannot replace TatAd or TatAy for protein translocation in B. subtilis, where it was shown to assist protein translocation by TatAyCy [75].
6 TatA and TatA/BC Complexes
While the Tat system can handle cargo proteins of up to 150 kDa [91], the Tat components are much smaller. This implies that they need to assemble into larger complexes that can facilitate membrane passage of larger cargo proteins [92]. Indeed, two types of Tat complexes were identified, namely TatA(B)C and TatA. In E. coli and thylakoids, membrane-embedded TatBC complexes are believed to bind cargo proteins, whereas recruitment of TatA complexes is required to facilitate their membrane passage [93,94,95]. In B. subtilis, the cargo receptor function of TatBC complexes is fulfilled by TatAdCd or TatAyCy complexes. Notably, the TatA complexes by themselves, especially those of B. subtilis (Table 1), are too small and homogeneous to allow passage of most cargo proteins [68, 77, 78]. The TatA-TatA/BC assemblies are thought to disassemble upon completed cargo translocation [96].
As shown by cross-linking studies, within TatBC complexes, TatC is first to interact with the N-region of a twin-arginine signal peptide [94, 97, 98]. Subsequently, deep insertion of the signal peptide into TatC will follow, leading to interaction of the H-domain with the transmembrane segment of TatB [54, 94, 99]. In turn, this leads to exposure of the signal peptidase cleave site in the C-region to signal peptidase on the trans-side of the membrane [63, 100,101,102]. Intriguingly, several lines of evidence, suggest that more than one cargo protein can be bound by assemblies of seven TatBC copies [60, 103,104,105]. Within these TatBC assemblies, TatC monomers have two TatB contact sites [61, 99, 102, 106, 107]. Further, the transmembrane segment of TatB appears to be positioned close to the site where translocase oligomerization is initiated by TatA, which suggests that TatB could serve as a regulatory surrogate of TatA [108,109,110]. The latter would be in line with the fact that TatB is absent from minimal TatAC translocases as encountered in B. subtilis. Furthermore, cross-linking analyses show that cargo docking via the signal peptide leads to conformational changes that rearrange TatC’s binding site for TatA and TatB [52]. Binding of a signal peptide changes the arrangement of TatC from head-to-tail to tail-to-tail [106].
TatBC complexes contain small amounts of TatA that may serve as points of TatA nucleation for forming the active translocase [111, 112]. Most TatA molecules are, however, present in TatA complexes. The TatA complexes of E. coli are very heterogeneous, ranging from 100 to 500 kDa with intermediate size intervals of ~ 34 kDa [48, 54, 113,114,115]. In contrast, TatAc, TatAd and TatAy complexes in Bacillus are much smaller with molecular masses of ~ 100, ~ 270 and ~ 200 kDa, respectively [76, 77].
7 Tat Translocation Mechanism
Despite almost three decades of research, the Tat translocation mechanism is still incompletely understood. As outlined above, cargo translocation is initiated at TatA/BC complexes and then facilitated by TatA [113]. This may involve either pore formation [116] or membrane weakening [43, 117].
Based on low-resolution EM images, it was proposed that TatA complexes have a pore of 8.5–13 nm that might accommodate cargo proteins of varying size [116, 118]. This pore would be closed by a lid at the cytoplasmic side membrane, resembling a ‘trap door’, which could swing open with the help of a conserved Gly residue in the hinge region of TatA to allow cargo passage [118, 119]. In this scenario, after cargo docking onto TatBC, TatA would be recruited to form an oligomeric ring conforming to the size of the cargo protein [120, 121]. Although this model appears attractive, the trap door concept has not been confirmed in other studies [46, 48, 122]. Moreover, complexes of the TatA paralogue TatE (50–110 kDa) appear too small for pore formation [43].
More recently, it was proposed that TatA complexes might serve to weaken the membrane [48, 106, 117, 123]. This would relate to the relatively short transmembrane domain of TatA that can locally restrict the membrane thickness. This membrane weakening would only occur upon cargo binding and interaction of the mature part of the cargo protein with the amphipathic helix of TatA [94, 99, 123,124,125,126]. In the absence of cargo, the membrane weakening would not take place as immersion of the amphipathic helix of TatA in the membrane would preserve the membrane integrity, as was shown for the thylakoidal TatA [122].
As mentioned above, protein translocation via Tat is exclusively driven by the proton-motive force, which consists of the ΔpH and the electric potential Δψ across the membrane [127]. Early studies into the energetics of Tat were performed in vitro with the plant thylakoid system. In the presence of light and a ΔpH, but in the absence of nucleotides, the photosystem II oxygen-evolving Tat cargo protein tOE23 was still exported [128]. In addition, a phage shock protein PspA, involved in maintaining the proton-motive force, was found to increase Tat translocation in bacteria [129]. However, in vivo studies in the green alga Chlamydomonas reinhardtii showed that the system can still transport proteins without a thylakoidal ΔpH, which can be explained by the fact that the Tat pathway can use both the ΔpH and Δψ [130, 131]. As a consequence of this equivalency, an antiporter mechanism was suggested where a coupling of H+ flow and protein transport has been suggested [132]. Of note, the counterflow of protons necessary for Tat protein export was estimated to amount about 7.9 × 104 protons per molecule [133]. This is equivalent to 10,000 ATP molecules, 3% of the energy produced by a chloroplast, so it is a considerable cost to the cell.
With regards to individual steps of the translocation mechanism, in vitro studies have shown that the proton-motive force is not required for protein targeting or protein binding to TatBC, but for the more advanced binding stages and oligomerisation of TatA [94, 134]. For thylakoids it was proposed that the ΔpH could potentially protonate TatA (Tha4) at the Glu10 residue, making it energetically feasible to move up in the membrane to its docking site in TatC (Gln234) [112]. However, in an earlier study this Glu10 residue was replaced with Gln, as well as with Ala or Asp, and all of these changes severely reduced the ability of TatA to facilitate protein transport [135]. While this shows the importance of the Glu10 residue and a negative charge at this position for translocation activity, it is not certain whether this implies a role of Glu10 in sensing the thylakoidal luminal pH through protonation, or whether Glu10 forms a salt bridge with a basic residue somewhere else [135]. It is also still unclear how the assembly of TatA in E. coli is facilitated by the proton-motive force, as it has been shown through in vitro studies that the transport driving force is largely provided by the Δψ [136]. In fact, these studies indicate that two distinct Δψ-dependent steps drive protein transport: a first step would involve a ∆ψ of relatively high magnitude that may be short-lived, and a second step of longer duration would require a ∆ψ of relatively low magnitude. When the ∆ψ was increased, so was the transport speed [94, 136]. This raises the question, how exactly the ∆ψ drives protein transport via Tat in E. coli and why this is apparently different in thylakoids, where the ΔpH represents the driving force for protein transport. A conceivable scenario is that movement of certain charged regions within the membrane-embedded E. coli Tat proteins could be induced by a ∆ψ, whereas this process would be induced by the ΔpH in the chloroplast thylakoidal membrane. Altogether, it is presently still unclear whether a potential across the membrane drives charge movements or whether proton transport by Tat takes place.
8 Chaperoning of Tat Cargo Proteins
One of the major hallmarks of the Tat pathway is its ability to selectively transport fully folded cofactor-containing proteins. To this end, the system involves different mechanisms. Translocation of particular cargo proteins requires the aid of dedicated chaperones, known as redox enzyme maturation proteins (REMPs) [137, 138]. An example of a Tat cargo protein involving a REMP for export is the oxidoreductase trimethylamine-N-oxide (TMAO) reductase (TorA; Fig. 1). This enzyme is encoded by the torCAD operon, where torA encodes the TMAO reductase, torC its haem-binding quinol oxidase and torD its REMP. In particular, TorD recognizes and binds the h- and c-regions of the TorA signal peptide most likely as a dimer [139,140,141]. Following signal peptide binding, TorD guides TorA export via Tat in a GTP-dependent manner. In this scenario, the affinity of TorD for GTP increases upon signal peptide binding, and GTP presumably controls cycles of signal peptide binding and release of TorD, thereby preventing premature protein degradation, coordinating cofactor assembly and foreseeing other maturation steps, such as membrane targeting and interaction [139, 140]. This coordination occurs until the pre-protein interacts with the Tat machinery.
9 Proofreading of the Folding State of Tat Cargo
The proofreading exhibited by the E. coli and B. subtilis Tat pathways is highly stringent to ensure misfolded proteins are not exported. Thus, the Tat complex rejects and may sometimes even degrade cargo proteins within the cytosol, although such degradation may also occur independently of the Tat system [142,143,144]. To note, the thylakoidal Tat system seems to have a less stringent ‘proofreading’ system as unfolded proteins are also imported [37].
To explore mechanisms of Tat proofreading, particular attention has been attributed to cofactor insertion. The native E. coli Tat cargo proteins NrfC and NapG were mutated to prevent their central cofactor FeS binding. Indeed this alteration blocked export [143]. The B. subtilis Rieske iron-sulphur cluster protein QcrA was also mutated to either stop cofactor binding or disulphide bond formation [145]. Here, a proofreading hierarchy was uncovered: mutant’s defective in disulphide bonding were quickly degraded, whereas those defective in cofactor binding accumulated in the cytoplasm and membrane. Two heme-binding proteins have also been investigated for proofreading. First, cytochrome C was shown to require heme insertion for export [146]. Subsequently, proofreading was investigated with the synthetic BT6 maquette protein, which binds two hemes and is Tat-dependently secreted in E. coli when provided with a TorA signal peptide [147]. His residues in BT6 were replaced with Ala to prevent heme binding. This showed that export was completely blocked if heme binding was completely prevented. Binding of one heme by BT6 allowed some export, whereas good export was observed when two hemes were bound [147]. Altogether, these findings suggest that Tat can somehow sense a protein’s conformational stability.
The requirement for conformational stability was further studied in vivo and in vitro with non-native Tat cargo proteins, such as E. coli PhoA and scFv or Fab antibody fragments. Export of these proteins only occurred in oxidizing conditions allowing disulphide bond formation prior to their interaction with the Tat machinery [40]. Nevertheless, some proteins provided with a twin-arginine signal peptide, like human growth hormone (hGH), scFv’s and interferon α2b, were exported to the periplasm without their disulphide bonds formed [148]. For hGH it was shown that this protein can form a near native state in absence of its two disulphide bonds. This is reminiscent of observations on the CueO protein of E. coli, which can still be exported via Tat without its bound copper cofactor. This probably relates to the fact that CueO without bound copper is structurally close to identical to CueO with bound copper [149].
Several studies in both bacteria and plants have used varying lengths of FG repeats from the yeast nuclear pore protein Nsp1p to probe the structural constraints for Tat-dependent export. These repeats intrinsically lack structure and are hydrophilic. Fused to a Tat signal peptide, export studies demonstrated that with increasing protein length, the translocation efficiency decreased: segments of 100–120 amino acids were tolerated, but a short hydrophobic stretch stopped export [150, 151]. Unstructured linkers were also placed between the signal peptide and the N-terminus of a mature Tat cargo protein and, surprisingly, an unstructured linker length of 110 amino acids was exported [152]. These findings imply that, despite the generally strict folding requirement for Tat cargo proteins, short unstructured polypeptide regions can be tolerated in particular protein contexts.
A recent study used scFv mutants [153], which were structurally defined, to identify what the E. coli Tat machinery recognizes as ‘unfolded’ and rejects for export [154]. Tat tolerated significant changes in hydrophobicity and charge, but did not export the scFv with an unstructured tail or without cytoplasmic disulphide bond formation via the so-called CyDisCo system. CyDisCo comprises the yeast mitochondrial thiol oxidase Erv1p plus the human protein disulfide isomerase PDI that, together, confer the ability to catalyse cytoplasmic disulphide bond formation.
Since it is still unclear what exactly the Tat complex rejects as misfolded, a key question is how the Tat complex rejects certain proteins. Tat proteins, misfolded or not, both interact with the Tat translocase. For example the PhoA protein provided with a twin-arginine signal peptide has been co-purified with TatBC [41]. This gave rise to the idea Tat does not innately have an inbuilt ‘proofreading’ mechanism, but rather an efficient degradation system that clears the Tat translocase. Indeed, the B. subtilis protease WprA was shown to interact directly with the Tat machinery and to be essential for protein secretion via TatAyCy [155, 156].
Lastly, in vitro site-specific photo cross-linking experiments revealed that unfolded TorA-PhoA associated with the Tat translocase, and that the interaction with the TatBC receptor site was perturbed as if the cargo was not correctly inserted into the binding socket [157]. This invoked the TatBC complex in proofreading of the cargo protein. This view is consistent with the identification of so-called quality control suppression (QCS) mutations within E. coli TatABC, which gave rise to less stringent proofreading [158]. The majority of these QCS mutations were confined to the unstructured or loop regions of TatABC, showing that proofreading at some level is undertaken by the Tat translocon.
10 Conclusion
In recent years, the core components of the Tat protein translocation systems have been identified, biochemically characterized and structurally defined. Yet, the precise mechanism by which Tat translocates proteins across the bacterial cytoplasmic membrane is still elusive due to the fact that high-resolution structural data of protein-translocating Tat complexes is currently missing. It can be anticipated that with the advent of novel high-resolution techniques for structural analyses of large protein complexes many of the so far unanswered fundamental questions in the Tat field can be tackled and answered.
References
Holland IB (2004) Translocation of bacterial proteins—an overview. Biochim Biophys Acta Mol Cell Res 1694:5–16
Palmer T, Sargent F, Berks BC (2010) The Tat protein export pathway. EcoSal Plus. https://doi.org/10.1128/ecosalplus.4.3.2
von Heijne G (1990) The signal peptide. J Membr Biol 115:195–201
Lüke I, Handford JI, Palmer T, Sargent F (2009) Proteolytic processing of Escherichia coli twin-arginine signal peptides by LepB. Arch Microbiol 191:919–925
Jongbloed JDH, Grieger U, Antelmann H et al (2004) Two minimal Tat translocases in Bacillus. Mol Microbiol 54:1319–1325
Dalbey RE, Wang P, van Dijl JM (2012) Membrane proteases in the bacterial protein secretion and quality control pathway. Microbiol Mol Biol Rev 76:311–330
Sakaguchi M, Tomiyoshi R, Kuroiwa T et al (1992) Functions of signal and signal-anchor sequences are determined by the balance between the hydrophobic segment and the N-terminal charge. Proc Natl Acad Sci 89:16–19
Berks BC (1996) A common export pathway for proteins binding complex redox cofactors? Mol Microbiol 22:393–404
Sargent F, Bogsch EG, Stanley NR et al (1998) Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J 17:3640–3650
Berks BC, Palmer T, Sargent F (2003) The Tat protein translocation pathway and its role in microbial physiology. Adv Microb Physiol 47:187–254
Stanley NR, Palmer T, Berks BC (2000) The twin arginine consensus motif of Tat signal peptides is involved in Sec-independent protein targeting in Escherichia coli. J Biol Chem 275:11591–11596
Buchanan G, Sargent F, Berks BC, Palmer T (2002) A genetic screen for suppressors of Escherichia coli Tat signal peptide mutations establishes a critical role for the second arginine within the twin-arginine motif. Arch Microbiol 177:107–112
Chaddock AM, Mant A, Karnauchov I et al (1995) A new type of signal peptide: central role of a twin-arginine motif in transfer signals for the delta pH-dependent thylakoidal protein translocase. EMBO J 14:2715–2722
Halbig D, Hou B, Freudl R et al (1999) Bacterial proteins carrying twin-R signal peptides are specifically targeted by the ΔpH-dependent transport machinery of the thylakoid membrane system. FEBS Lett 447:95–98
DeLisa MP, Samuelson P, Palmer T, Georgiou G (2002) Genetic analysis of the twin arginine translocator secretion pathway in bacteria. J Biol Chem 277:29825–29831
Hinsley AP, Stanley NR, Palmer T, Berks BC (2001) A naturally occurring bacterial Tat signal peptide lacking one of the “invariant” arginine residues of the consensus targeting motif. FEBS Lett 497:45–49
Kipping M, Lilie H, Lindenstrauß U et al (2003) Structural studies on a twin-arginine signal sequence. FEBS Lett 550:18–22
Cristóbal S, De Gier JW, Nielsen H, Von Heijne G (1999) Competition between Sec- and TAT-dependent protein translocation in Escherichia coli. EMBO J 18:2982–2990
Bogsch E, Brink S, Robinson C (1997) Pathway specificity for a ΔpH-dependent precursor thylakoid lumen protein is governed by a “Sec-avoidance” motif in the transfer peptide and a “Sec-incompatible” mature protein. EMBO J 16:3851–3859
Tullman-ercek D, Delisa MP, Kawarasaki Y et al (2009) NIH Public Access 282:8309–8316
Mould RM, Robinson C (1991) A proton gradient is required for the transport of two lumenal oxygen-evolving proteins across the thylakoid membrane. J Biol Chem 266:12189–12193
Klösgen RB, Brock IW, Herrmann RG, Robinson C (1992) Proton gradient-driven import of the 16 kDa oxygen-evolving complex protein as the full precursor protein by isolated thylakoids. Plant Mol Biol 18:1031–1034
Clark SA, Theg SM (1997) A folded protein can be transported across the chloroplast envelope and thylakoid membranes. Mol Biol Cell 8:923–934
Mori H, Summer EJ, Ma X, Cline K (1999) Component specificity for the thylakoidal Sec and ΔpH-dependent protein transport pathways. J Cell Biol 146:45–55
Settles AM, Yonetani A, Baron A et al (1997) Sec-independent protein translocation by the maize Hcf106 protein. Science 278:1467–1470
Cline K, Mori H (2001) Thylakoid ΔpH-dependent precursor proteins bind to a cpTatC-Hcf106 complex before Tha4-dependent transport. J Cell Biol 154:719–729
Wu LF, Ize B, Chanal A, Quentin Y, Fichant G (2000) Bacterial twin-arginine signal peptide-dependent protein translocation pathway: evolution and mechanism. J Mol Microbiol Biotechnol 2:179–189
Yen MR, Tseng YH, Nguyen EH et al (2002) Sequence and phylogenetic analyses of the twin-arginine targeting (Tat) protein export system. Arch Microbiol 177:441–450
Bogsch EG, Sargent F, Stanley NR et al (1998) An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J Biol Chem 273:18003–18006
Sargent F, Stanley NR, Berks BC, Palmer T (1999) Sec-independent protein translocation in Escherichia coli. A distinct and pivotal role for the TatB protein. J Biol Chem 274:36073–36082
Weiner JH, Bilous PT, Shaw GM et al (1998) A novel and ubiquitous system for membrane targeting and secretion of cofactor-containing proteins. Cell 93:93–101
Rodrigue A, Chanal A, Beck B, Muller M, Wu LF (1999) Co-translocation of a periplasmic enzyme complex by a hitchhiker mechanism through the bacterial tat pathway. J Biol Chem 274:13223–13228
Palmer T, Sargent F, Berks BC (2005) Export of complex cofactor-containing proteins by the bacterial Tat pathway. Trends Microbiol 13:175–180
Bernhardt TG, De Boer PAJ (2003) The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol Microbiol 48:1171–1182
Ize B, Stanley NR, Buchanan G, Palmer T (2003) Role of the Escherichia coli Tat pathway in outer membrane integrity. Mol Microbiol 48:1183–1193
Ding Z, Christie PJ (2003) Agrobacterium tumefaciens twin-arginine-dependent translocation is important for virulence, flagellation, and chemotaxis but not type IV secretion. J Bacteriol 185:760–771
Hynds PJ, Robinson D, Robinson C (1998) The Sec-independent twin-arginine translocation system can transport both tightly folded and malfolded proteins across the thylakoid membrane. J Biol Chem 273:34868–34874
Santini CL, Bernadac A, Zhang M et al (2001) Translocation of jellyfish green fluorescent protein via the Tat system of Escherichia coli and change of its periplasmic localization in response to osmotic up-shock. J Biol Chem 276:8159–8164
Alanen HI, Walker KL, Lourdes Velez Suberbie M et al (2014) Efficient export of human growth hormone, interferon α2b and antibody fragments to the periplasm by the Escherichia coli Tat pathway in the absence of prior disulfide bond formation. Biochim Biophys Acta Mol Cell Res 1853:756–763
Delisa MP, Tullman D, Georgiou G (2003) Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. PNAS 100:6115–6120
Richter S, Brüser T (2005) Targeting of unfolded PhoA to the TAT translocon of Escherichia coli. J Biol Chem 280:42723–42730
Jack RL, Sargent F, Berks BC et al (2001) Constitutive expression of Escherichia coli tat genes indicates an important role for the twin-arginine translocase during aerobic and anaerobic growth. J Bacteriol 183:1801–1804
Baglieri J, Beck D, Vasisht N et al (2012) Structure of TatA paralog, TatE, suggests a structurally homogeneous form of Tat protein translocase that transports folded proteins of differing diameter. J Biol Chem 287:7335–7344
Stanley NR, Findlay K, Berks BC, Palmer T (2001) Escherichia coli strains blocked in Tat-dependent protein export exhibit pleiotropic defects in the cell envelope. J Bacteriol 183:139–144
Sargent F, Gohlke U, De Leeuw E et al (2001) Purified components of the Escherichia coli Tat protein transport system form a double-layered ring structure. Eur J Biochem 268:3361–3367
Koch S, Fritsch MJ, Buchanan G, Palmer T (2012) Escherichia coli TatA and TatB proteins have N-out, C-in topology in intact cells. J Biol Chem 287:14420–14431
Hu Y, Zhao E, Li H et al (2010) Solution NMR structure of the TatA component of the twin-arginine protein transport system from gram-positive bacterium Bacillus subtilis. J Am Chem Soc 132:15942–15944
Rodriguez F, Rouse SL, Tait CE et al (2013) Structural model for the protein-translocating element of the twin-arginine transport system. Proc Natl Acad Sci USA 110:E1092–E1101
Barrett CML, Ray N, Thomas JD et al (2003) Quantitative export of a reporter protein, GFP, by the twin-arginine translocation pathway in Escherichia coli. Biochem Biophys Res Commun 304:279–284
Hicks MG, De Leeuw E, Porcelli I et al (2003) The Escherichia coli twin-arginine translocase: conserved residues of TatA and TatB family components involved in protein transport. FEBS Lett 539:61–67
Greene NP, Porcelli I, Buchanan G et al (2007) Cysteine scanning mutagenesis and disulfide mapping studies of the TatA component of the bacterial twin arginine translocase. J Biol Chem 282:23937–23945
Eimer E, Fröbel J, Blümmel AS, Müller M (2015) TatE as a regular constituent of bacterial twin-arginine protein translocases. J Biol Chem 290:29281–29289
Eimer E, Kao WC, Fröbel J et al (2018) Unanticipated functional diversity among the TatA-type components of the Tat protein translocase. Sci Rep 8:1–12
Zhang Y, Wang L, Hu Y, Jin C (2014) Solution structure of the TatB component of the twin-arginine translocation system. Biochim Biophys Acta Biomembr 1838:1881–1888
Lee PA, Buchanan G, Stanley NR et al (2002) Truncation analysis of TatA and TatB defines the minimal functional units required for protein translocation. J Bacteriol 184:5871–5879
Barrett CML, Mathers JE, Robinson C (2003) Identification of key regions within the Escherichia coli TatAB subunits. FEBS Lett 537:42–46
Blaudeck N, Kreutzenbeck P, Müller M et al (2005) Isolation and characterization of bifunctional Escherichia coli TatA mutant proteins that allow efficient Tat-dependent protein translocation in the absence of TatB. J Biol Chem 280:3426–3432
Barrett CML, Freudl R, Robinson C (2007) Twin arginine translocation (Tat)-dependent export in the apparent absence of TatABC or TatA complexes using modified Escherichia coli TatA subunits that substitute for TatB. J Biol Chem 282:36206–36213
Jongbloed JDH, Van Der Ploeg R, Van Dijl JM (2006) Bifunctional TatA subunits in minimal Tat protein translocases [2]. Trends Microbiol 14:2–4
Tarry MJ, Schäfer E, Chen S et al (2009) Structural analysis of substrate binding by the TatBC component of the twin-arginine protein transport system. Proc Natl Acad Sci USA 106:13284–13289
Kneuper H, Maldonado B, Jäger F et al (2012) Molecular dissection of TatC defines critical regions essential for protein transport and a TatB-TatC contact site. Mol Microbiol 85:945–961
Behrendt J, Standar K, Lindenstrauß U, Brüser T (2004) Topological studies on the twin-arginine translocase component TatC. FEMS Microbiol Lett 234:303–308
Ramasamy S, Abrol R, Suloway CJM, Clemons WM (2013) The glove-like structure of the conserved membrane protein TatC provides insight into signal sequence recognition in twin-arginine translocation. Structure 21:777–788
Rollauer SE, Tarry MJ, Graham JE, Jääskeläinen M, Jäger F, Johnson S, Krehenbrink M, Liu SM, Lukey MJ, Marcoux J, McDowell MA, Rodriguez F, Roversi P, Stansfeld PJ, Robinson CV, Sansom MS, Palmer T, Högbom M, Berks BC, Lea SM (2012) Structure of the TatC core of the twin-arginine protein transport system. Nature 492(7428):210–214. https://doi.org/10.1038/nature11683
Blümmel AS, Drepper F, Knapp B et al (2017) Structural features of the TatC membrane protein that determine docking and insertion of a twin-arginine signal peptide. J Biol Chem 292:21320–21329
Holzapfel E, Eisner G, Alami M et al (2007) The entire N-terminal half of TatC is involved in twin-arginine precursor binding. Biochemistry 46:2892–2898
Goosens VJ, Monteferrante CG, Van Dijl JM (2014) The Tat system of Gram-positive bacteria. Biochim Biophys Acta Mol Cell Res 1843:1698–1706
Beck D, Vasisht N, Baglieri J et al (2009) Subcellular localization of TatAd of Bacillus subtilis depends on the presence of TatCd or TatCy. J Biol Chem 191:4410–4418
Schaerlaekens K, Schierová M, Lammertyn E et al (2001) Twin-arginine translocation pathway in Streptomyces lividans. J Bacteriol 183:6727–6732
Jongbloed JDH, Martin U, Antelmann H et al (2000) TatC is a specificity determinant for protein secretion via the twin-arginine translocation pathway. J Biol Chem 275:41350–41357
van der Ploeg R, Mäder U, Homuth G et al (2011) Environmental salinity determines the specificity and need for tat-dependent secretion of the YwbN protein in Bacillus subtilis. PLoS ONE 6:18140
Monteferrante CG, Miethke M, Van Der Ploeg R et al (2012) Specific targeting of the metallophosphoesterase YkuE to the Bacillus cell wall requires the twin-arginine translocation system. J Biol Chem 287:29789–29800
Miethke M, Monteferrante CG, Marahiel MA, van Dijl JM (2013) The Bacillus subtilis EfeUOB transporter is essential for high-affinity acquisition of ferrous and ferric iron. Biochim Biophys Acta Mol Cell Res 1833:2267–2278
Goosens VJ, Otto A, Glasner C et al (2013) Novel twin-arginine translocation pathway-dependent phenotypes of Bacillus subtilis unveiled by quantitative proteomics. J Proteome Res 12:796–807
Goosens VJ, De-San-Eustaquio-Campillo A, Carballido-López R, van Dijl JM (2015) A Tat ménage à trois—the role of Bacillus subtilis TatAc in twin-arginine protein translocation. Biochim Biophys Acta Mol Cell Res 1853:2745–2753
Barnett JP, Eijlander RT, Kuipers OP, Robinson C (2008) A minimal tat system from a gram-positive organism: a bifunctional TatA subunit participates in discrete TatAC and TatA complexes. J Biol Chem 283:2534–2542
Monteferrante CG, Baglieri J, Robinson C, van Dijl JM (2012) TatAc, the third TatA subunit of Bacillus subtilis, can form active twin-arginine translocases with the TatCd and TatCy subunits. Appl Environ Microbiol 78:4999–5001
Beck D, Vasisht N, Baglieri J et al (2013) Ultrastructural characterisation of Bacillus subtilis TatA complexes suggests they are too small to form homooligomeric translocation pores. Biochim Biophys Acta Mol Cell Res 1833:1811–1819
Porcelli I, De Leeuw E, Wallis R et al (2002) Characterization and membrane assembly of the TatA component of the Escherichia coli twin-arginine protein transport system. Biochemistry 41:13690–13697
Müller SD, De Angelis AA, Walther TH et al (2007) Structural characterization of the pore forming protein TatAd of the twin-arginine translocase in membranes by solid-state 15 N-NMR. Biochim Biophys Acta Biomembr 1768:3071–3079
Walther TH, Grage SL, Roth N, Ulrich AS (2010) Membrane alignment of the pore-forming component TatAd of the twin-arginine translocase from Bacillus subtilis resolved by solid-state NMR spectroscopy. J Am Chem Soc 132:15945–15956
Lange C, Müller SD, Walther TH et al (2007) Structure analysis of the protein translocating channel TatA in membranes using a multi-construct approach. Biochim Biophys Acta Biomembr 1768:2627–2634
Barnett JP, Lawrence J, Mendel S, Robinson C (2011) Expression of the bifunctional Bacillus subtilis TatAd protein in Escherichia coli reveals distinct TatA/B-family and TatB-specific domains. Arch Microbiol 193:583–594
Schreiber S, Stengel R, Westermann M et al (2006) Affinity of TatCdfor TatA delucidates its receptor function in the Bacillus subtilis twin arginine translocation (Tat) translocase system. J Biol Chem 281:19977–19984
Nolandt OV, Walther TH, Roth S et al (2009) Structure analysis of the membrane protein TatCd from the Tat system of B. subtilis by circular dichroism. Biochim Biophys Acta Biomembr 1788:2238–2244
Van Der Ploeg R, Barnett JP, Vasisht N et al (2011) Salt sensitivity of minimal twin arginine translocases. J Biol Chem 286:43759–43770
Goosens VJ, van Dijl JM (2017) Twin-arginine protein translocation. Curr Top Microbiol Imunol 404:69–94
Patel R, Vasilev C, Beck D et al (2014) A mutation leading to super-assembly of twin-arginine translocase (Tat) protein complexes. Biochim Biophys Acta Mol Cell Res 1843:1978–1986
Eijlander RT, Jongbloed JDH, Kuipers OP (2009) Relaxed specificity of the Bacillus subtilis TatAdCd translocase in Tat-dependent protein secretion. J Bacteriol 91:196–202
Eijlander RT, Kolbusz MA, Berendsen EM, Kuipers OP (2009) Effects of altered TatC proteins on protein secretion efficiency via the twin-arginine translocation pathway of Bacillus subtilis. Microbiology 155:1776–1785
Berks BC, Sargent F, Palmer T (2000) The Tat protein export pathway. Mol Microbiol 35:260–274
Rose P, Fröbel J, Graumann PL, Müller M (2013) Substrate-dependent assembly of the Tat translocase as observed in live Escherichia coli cells. PLoS ONE 8:1–17
Berks BC, Lea SM, Stansfeld PJ (2014) Structural biology of tat protein transport. Curr Opin Struct Biol 27:32–37
Alami M, Lüke I, Deitermann S et al (2003) Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol Cell 12:937–946
Celedon JM, Cline K (2012) Stoichiometry for binding and transport by the twin Arginine translocation system. J Cell Biol 197:523–534
Alcock F, Baker MAB, Greene NP et al (2013) Live cell imaging shows reversible assembly of the TatA component of the twin-arginine protein transport system. Proc Natl Acad Sci 110:E3650–E3659
Gérard F, Cline K (2006) Efficient twin arginine translocation (Tat) pathway transport of a precursor protein covalently anchored to its initial cpTatC binding site. J Biol Chem 281:6130–6135
Kreutzenbeck P, Kröger C, Lausberg F et al (2007) Escherichia coli twin arginine (Tat) mutant translocases possessing relaxed signal peptide recognition specificities. J Biol Chem 282:7903–7911
Maurer C, Panahandeh S, Jungkamp A-C et al (2010) TatB functions as an oligomeric binding site for folded Tat precursor proteins. Mol Biol Cell 21:4151–4161
Zoufaly S, Fröbel J, Rose P et al (2012) Mapping precursor-binding site on TatC subunit of twin arginine-specific protein translocase by site-specific photo cross-linking. J Biol Chem 287:13430–13441
Fröbel J, Rose P, Lausberg F et al (2012) Transmembrane insertion of twin-arginine signal peptides is driven by TatC and regulated by TatB. Nat Commun 3:1311
Blümmel AS, Haag LA, Eimer E et al (2015) Initial assembly steps of a translocase for folded proteins. Nat Commun 6:7234
Bolhuis A, Mathers JE, Thomas JD et al (2001) TatB and TatC form a functional and structural unit of the twin-arginine translocase from Escherichia coli. J Biol Chem 276:20213–20219
Behrendt J, Brüser T (2014) The TatBC complex of the tat protein translocase in Escherichia coli and its transition to the substrate-bound TatABC complex. Biochemistry 53:2344–2354
Ma X, Cline K (2010) Multiple precursor proteins bind individual Tat receptor complexes and are collectively transported. EMBO J 29:1477–1488
Rollauer SE, Tarry MJ, Graham JE et al (2012) Structure of the TatC core of the twin-arginine protein transport system. Nature 492:210–214
Lee PA, Orriss GL, Buchanan G et al (2006) Cysteine-scanning mutagenesis and disulfide mapping studies of the conserved domain of the twin-arginine translocase TatB component. J Biol Chem 281:34072–34085
Cline K (2015) Mechanistic aspects of folded protein transport by the twin arginine translocase (Tat). J Biol Chem 290:16530–16538
Alcock F, Stansfeld PJ, Basit H et al (2016) Assembling the tat protein translocase. Elife 5:e20718
Habersetzer J, Moore K, Cherry J et al (2017) Substrate-triggered position switching of TatA and TatB during Tat transport in Escherichia coli. Open Biol 7:8
Orriss GL, Tarry MJ, Ize B et al (2007) TatBC, TatB, and TatC form structurally autonomous units within the twin arginine protein transport system of Escherichia coli. FEBS Lett 581:4091–4097
Aldridge C, Ma X, Gerard F, Cline K (2014) Substrate-gated docking of pore subunit Tha4 in the TatC cavity initiates Tat translocase assembly. J Cell Biol 205:51–65
Hauer RS, Schlesier R, Heilmann K et al (2013) Enough is enough: TatA demand during Tat-dependent protein transport. Biochim Biophys Acta Mol Cell Res 1833:957–965
Oates J, Barrett CML, Barnett JP et al (2005) The Escherichia coli twin-arginine translocation apparatus incorporates a distinct form of TatABC complex, spectrum of modular TatA complexes and minor TatAB complex. J Mol Biol 346:295–305
White GF, Schermann SM, Bradley J et al (2010) Subunit organization in the TatA complex of the twin arginine protein translocase: a site-directed EPR spin labeling study. J Biol Chem 285:2294–2301
Gohlke U, Pullan L, McDevitt CA et al (2005) The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Natl Acad Sci USA 102:10482–10486
Brüser T, Sanders C (2003) An alternative model of the twin arginine translocation system. Microbiol Res 158:7–17
Gouffi K, Gérard F, Santini CL, Wu LF (2004) Dual topology of the Escherichia coli TatA protein. J Biol Chem 279:11608–11615
Walther TH, Gottselig C, Grage SL et al (2013) Folding and self-assembly of the TatA translocation pore based on a charge zipper mechanism. Cell 152:316–326
Dabney-Smith C, Mori H, Cline K (2006) Oligomers of Tha4 organize at the thylakoid Tat translocase during protein transport. J Biol Chem 281:5476–5483
Chan CS, Zlomislic MR, Tieleman DP, Turner RJ (2007) The TatA subunit of Escherichia coli twin-arginine translocase has an N-in topology. Biochemistry 46:7396–7404
Aldridge C, Storm A, Cline K, Dabney-Smith C (2012) The chloroplast twin arginine transport (Tat) component, Tha4, undergoes conformational changes leading to Tat protein transport. J Biol Chem 287:34752–34763
Taubert J, Hou B, Risselada HJ et al (2015) TatBC-Independent TatA/Tat substrate interactions contribute to transport efficiency. PLoS ONE 10:1–24
Pal D, Fite K, Dabney-Smith C (2013) Direct interaction between a precursor mature domain and transport component Tha4 during twin arginine transport of chloroplasts. Plant Physiol 161:990–1001
Taubert J, Brüser T (2014) Twin-arginine translocation-arresting protein regions contact TatA and TatB. Biol Chem 395:827–836
Hou B, Heidrich ES, Mehner-Breitfeld D, Brüser T (2018) The TatA component of the twin-arginine translocation system locally weakens the cytoplasmic membrane of Escherichia coli upon protein substrate binding. J Biol Chem 293:7592–7605
Mould RM, Shackleton JB, Robinson C (1991) Transport of proteins into chloroplasts. Requirements for the efficient import of two lumenal oxygen-evolving complex proteins into isolated thylakoids. J Biol Chem 266:17286–17289
Mori H, Cline K (2002) A twin arginine signal peptide and the pH gradient trigger reversible assembly of the thylakoid ΔpH/Tat translocase. J Cell Biol 157:205–210
DeLisa MP, Lee P, Palmer T, Georgiou G (2004) Phage shock protein PspA of Escherichia coli relieves saturation of protein export via the Tat pathway. J Bacteriol 186:366–373
Finazzi G, Chasen C, Wollman FA, De Vitry C (2003) Thylakoid targeting of Tat passenger proteins shows no ΔpH dependence in vivo. EMBO J 22:807–815
Braun NA, Davis AW, Theg SM (2007) The chloroplast tat pathway utilizes the transmembrane electric potential as an energy source. Biophys J 93:1993–1998
Musser SM, Theg SM (2000) Proton transfer limits protein translocation rate by the thylakoid ΔpH/Tat machinery. Biochemistry 39:8228–8233
Alder NN, Theg SM (2003) Energetics of protein transport across biological membranes: a study of the thylakoid ΔpH-dependent/cpTat pathway. Cell 112:231–242
Gérard F, Cline K (2007) The thylakoid proton gradient promotes an advanced stage of signal peptide binding deep within the Tat pathway receptor complex. J Biol Chem 282:5263–5272
Dabney-Smith C, Mori H, Cline K (2003) Requirement of a Tha4-conserved transmembrane glutamate in thylakoid Tat translocase assembly revealed by biochemical complementation. J Biol Chem 278:43027–43033
Bageshwar UK, Musser SM (2007) Two electrical potential-dependent steps are required for transport by the Escherichia coli Tat machinery. J Cell Biol 179:87–99
Turner RJ, Papish AL, Sargent F (2004) Sequence analysis of bacterial redox enzyme maturation proteins (REMPs). Can J Microbiol 50:225–238
Chan CS, Chang L, Rommens KL, Turner RJ (2009) Differential interactions between tat-specific redox enzyme peptides and their chaperones. J Bacteriol 191:2091–2101
Jack RL, Buchanan G, Dubini A, Hatzixanthis K, Palmer T, Sargent F (2004) Coordinating assembly and export of complex bacterial proteins. EMBO J 23:3962–3972
Hatzixanthis K, Clarke TA, Oubrie A, Richardson DJ, Turner RJ, Sargent F (2005) Signal peptide-chaperone interactions on the twin-arginine protein transport pathway. Proc Natl Acad Sci USA 102:8460–8465
Tranier S, Mortier-Barrière I, Ilbert M et al (2002) Characterization and multiple molecular forms of TorD from Shewanella massilia, the putative chaperone of the molybdoenzyme TorA. Protein Sci 11:2148–2157
Halbig D, Wiegert T, Blaudeck N et al (1999) The efficient export of NADP-containing glucose-fructose oxidoreductase to the periplasm of Zymomonas mobilis depends both on an intact twin- arginine motif in the signal peptide and on the generation of a structural export signal induced by cofactor bind. Eur J Biochem 263:543–551
Matos CFRO, Robinson C, Di Cola A (2008) The Tat system proofreads FeS protein substrates and directly initiates the disposal of rejected molecules. EMBO J 27:2055–2063
Lindenstrauss U, Matos CF, Graubner W, Robinson C, Brüser T (2010) Malfolded recombinant Tat substrates are Tat-independently degraded in Escherichia coli. FEBS Lett 584:3644–3648
Goosens VJ, Monteferrante CG, Van Dijl JM (2014) Co-factor insertion and disulfide bond requirements for twin-Arginine translocase-dependent export of the Bacillus subtilis Rieske protein QcrA. J Biol Chem 289:13124–13131
Sanders C, Wethkamp N, Lill H (2001) Transport of cytochrome c derivatives by the bacterial Tat protein translocation system. Mol Microbiol 41:241–246
Sutherland GA, Grayson KJ, Adams NBP et al (2018) Probing the quality control mechanism of the Escherichia coli twin-arginine translocase with folding variants of a de novo—designed heme protein. J Biol Chem 293:6672–6681
Alanen HI, Walker KL, Lourdes Velez Suberbie M et al (2015) Efficient export of human growth hormone, interferon α2b and antibody fragments to the periplasm by the Escherichia coli Tat pathway in the absence of prior disulfide bond formation. Biochim Biophys Acta Mol Cell Res 1853:756–763
Stolle P, Hou B, Brüser T (2016) The Tat substrate CueO is transported in an incomplete folding state. J Biol Chem 291:13520–13528
Cline K, McCaffery M (2007) Evidence for a dynamic and transient pathway through the TAT protein transport machinery. EMBO J 26:3039–3049
Richter S, Lindenstrauss U, Lücke C et al (2007) Functional tat transport of unstructured, small, hydrophilic proteins. J Biol Chem 282:33257–33264
Lindenstrauß U, Brüser T (2009) Tat transport of linker-containing proteins in Escherichia coli. FEMS Microbiol Lett 295:135–140
Austerberry JI, Dajani R, Panova S et al (2017) The effect of charge mutations on the stability and aggregation of a human single chain Fv fragment. Eur J Pharm Biopharm 115:18–30
Jones AS, Austerberry JI, Dajani R et al (2016) Proofreading of substrate structure by the Twin-Arginine Translocase is highly dependent on substrate conformational flexibility but surprisingly tolerant of surface charge and hydrophobicity changes. Biochim Biophys Acta Mol Cell Res 1863:3116–3124
Monteferrante CG, Mackichan C, Marchadier E et al (2013) Mapping the twin-arginine protein translocation network of Bacillus subtilis. Proteomics 13:800–811
Krishnappa L, Monteferrante CG, van Dijl JM (2012) Degradation of the twin-arginine translocation substrate YwBn by extracytoplasmic proteases of Bacillus subtilis. Appl Environ Microbiol 78:7801–7804
Panahandeh S, Maurer C, Moser M et al (2008) Following the path of a twin-arginine precursor along the TatABC translocase of Escherichia coli. J Biol Chem 283:33267–33275
Rocco MA, Waraho-Zhmayev D, DeLisa MP (2012) Twin-arginine translocase mutations that suppress folding quality control and permit export of misfolded substrate proteins. Proc Natl Acad Sci 109:13392–13397
Barnett JP, Van Der Ploeg R, Eijlander RT, Nenninger A, Mendel S, Rozeboom R, Kuipers OP, Van Dijl JM, Robinson C (2009) The twin-arginine translocation (Tat) systems from Bacillus subtilis display a conserved mode of complex organization and similar substrate recognition requirements. FEBS J 276:232–243
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Frain, K.M., Robinson, C. & van Dijl, J.M. Transport of Folded Proteins by the Tat System. Protein J 38, 377–388 (2019). https://doi.org/10.1007/s10930-019-09859-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-019-09859-y