
Abstract
Reduced genetic diversity due to founder effects often is expected for invasive populations. The present study examined two nuclear gene regions and one mitochondrial gene to evaluate the origins and genetic diversity of Gemma gemma, a ‘stow-away’ that was introduced to California more than 100 years ago with the importation of the Eastern oyster, Crassostrea virginica, from the United States’ Atlantic coast. A previous investigation involving mitochondrial DNA cytochrome-c-oxidase subunit I sequences reported no significant difference in haplotype diversity between the native and introduced populations; however, estimates of allelic (or haplotypic) variability are insensitive to losses of rare alleles that may accompany founder events and population bottlenecks. Estimates of allele richness and the distribution of rare alleles provide more sensitive indicators of such events. The present investigation of introduced and potential source populations identified lower allele richness and number of singleton alleles in California samples. Atlantic coast Gemma exhibit a sharp phylogeographic transition between northeastern (New York through New England) and mid-Atlantic (southern New Jersey through Virginia) subpopulations, which appear latitudinally inverted for the California Gemma populations. These genetic results, and information from the transportation history of the Eastern oyster, help to clarify processes involved in the introduction of this invasive species.
Similar content being viewed by others

Introduction
Human travel and commerce during the past century have resulted in an increasing number of intentional and inadvertent introductions of species to locations where they previously did not occur (Ruiz et al. 2013). Understanding establishment processes, population dynamics, and the specific sources of these invasive species may aid in the development to control programs. Levels of genetic diversity in introduced populations are hypothesized to influence a species’ capacity to adapt to novel environments (Allendorf and Lundquist 2003). Genetic diversity is expected to increase with the number of propagules involved in the initial establishment (founder size) and the intrinsic rate of increase of an introduced population (Nei et al. 1975). Reduced diversity is commonly expected for founding populations (Roman and Darling 2007) when compared with the source populations; nonetheless, several recent studies reveal similar or even higher genetic diversity in introduced populations, particularly if introductions occurred multiple times and from geographically diverse sources (Brown and Stepien 2010; Gillis et al. 2009; Kolbe et al. 2004).
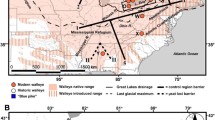
Native and invasive populations of the amethyst gem clam, Gemma gemma Totten 1834 (hereafter, Gemma), previously were compared for mitochondrial DNA diversities in their native and introduced ranges along the Atlantic and Pacific coasts of North America (Hoos et al. 2010). This ovoviviparous clam provides an unusual opportunity to examine genetic characteristics of an invasive species. Gemma is reported to live naturally in intertidal muds along the Atlantic coast from Labrador, Canada, to Texas in the Gulf of Mexico, and often is associated with oyster beds. Following completion of the Transcontinental Railroad in 1869, Gemma and other ‘stow-aways’ were inadvertently translocated to California coastal locations with plantings of the Eastern oyster, Crassostrea virginica (Carlton 1979; Ingersoll 1881; Kochiss 1974; Miller 2000). Hoos et al. (2010) briefly summarized the history of the Eastern oyster trade and exports. A detailed history of the California oyster industry is provided by Barrett (1963) and Miller (2000). The oysters and stow-aways were packed in wooden barrels and transported primarily from New York City to San Francisco, CA, through 1940. The exported oysters originated from multiple sources along the east coast. During the late eighteenth century and throughout the nineteenth century, ailing oyster stocks from the Delaware Bay and north to New England states were supplemented with ones dredged from Chesapeake Bay locations (Ingersoll 1881; Kochiss 1974; Miller 2000). Following a grow-out period, the explants were sold to New York dealers for direct consumption and export. A self-sustaining eastern oyster industry ultimately failed in California, but stow-aways such as Gemma, have thrived in several bays and lagoons in and adjacent to the San Francisco Bay.
To identify the primary source populations, Hoos et al. (2010) compared mitochondrial cytochrome c oxidase subunit I (COI) DNA diversity in Gemma samples from five California locations with native locations ranging from New England to North Carolina. Two California populations that corresponded to distinct Eastern populations were distinguished by their mtDNA haplotype frequencies. Curiously, the latitudinal distributions of the invasive populations appeared inverted with respect to the native populations. Southern mtDNA haplotypes indicative of Chesapeake Bay and North Carolina occurred in a northern group that invaded Bodega Harbor and Tomales Bay, whereas northern haplotypes indicative of Delaware Bay northward to Maine occur in a southern group that invaded Bolinas Lagoon, San Francisco Bay, and Elkhorn Sough. Oddly, Gemma sampled from several locations in San Francisco Bay, the principal recipient of Eastern oyster stocks, had among the lowest haplotypic diversities (h) for the California locations. Three secondary locations that received explants from San Francisco Bay (Tomales Bay, Bolinas Lagoon, and Elkhorn Slough) had haplotype diversities (h), a commonly reported of measure of mitochondrial variation, comparable to some East coast locations, but the increases in h appeared due to greater evenness of haplotypic frequencies in the invasive populations. In contrast, the Bodega Harbor Gemma appeared fixed for a single haplotype, but similar fixation of mitochondrial haplotypes also was found at several Eastern locations. MtDNA mutates relatively rapidly, and novel haplotypes might be subjected to selective sweeps or frequency changes due to founder events and population bottlenecks. What caused the historical shifts in haplotype frequencies in Bodega Harbor and San Francisco Bay Gemma populations remains unknown, but together these observations suggest that reconstructions of historical (150 year-old) origins for an invasive species from present-day frequencies of a single gene should be treated with caution. MtDNA is inherited separately in maternal lineages from nuclear DNA and frequently has a higher mutation rate in animals. Also introgression of mtDNA has been noted in a broad range of animal taxa (e.g. Bachtrog et al. 2006; Boratyński et al. 2011). As a molecular marker, mtDNA has been questioned for its accuracy in reflecting the recent histories of some populations and species (Galtier et al. 2009).
An examination of independently assorting nuclear genes provides a means to assess genetic inferences regarding Gemma introductions. For example, Hoos et al. (2010) reported that mtDNA diversity was, on average, not substantially reduced in the California Gemma population. Haplotypic diversity was estimated analogous to expected heterozygosity for the nuclear genes, H = 1 − Σp 2i , where p i is the frequency of the ith allele. Rare alleles contribute very little to estimates of H, which is influenced mostly by the evenness of allelic frequencies (Nei 1975). Estimates of allelic richness (A), or the total number of alleles, provide more sensitive indicators of founder events and recent bottlenecks in population size, because rare alleles are rapidly lost during such events (Allendorf 1986; Cornuet and Luikart 1996). With appropriate corrections for sample size, the distribution and abundance of rare alleles, including ‘singletons’ (alleles that are found just once among all samples) and of ‘private’ alleles (those occurring in one population sample) provide useful indices for elucidating recent population bottlenecks and founder events (Behar et al. 2004; Kalinowsky 2005; Neilson and Wilson 2005). In the present study of native and introduced Gemma populations, we examined heterozygosity and allelic richness for partial sequences of two independent nuclear genes (nDNAs). Genes encoding the enzymes adenine nucleotide translocase (ANT) and Histone-3 (H3) were selected from a panel of candidate loci that were previously screened for consistency of amplifications and the existence of scorable polymorphisms. Specifically, the present study addressed these questions: (1) Do native and introduced populations of Gemma differ in nDNA diversity? (2) Are diversity estimates from nDNA concordant with those from mtDNA? and (3) Are biogeographic patterns similar in the west and east coast populations?
Materials and methods
Sample collection
Gemma individuals were collected in 2005 from five west coast locations in California and seven east coast locations ranging from Maine to North Carolina as described in Hoos et al. 2010 (Table 1). DNA was extracted from soft tissues with DNAzol following the manufacturer’s protocol (Molecular Research Center Inc., Cincinnati, OH, USA), and preserved at −70 °C (Hoos et al. 2010). A 448 bp fragment of mtDNA COI was sequenced and analyzed in a previous study (Hoos et al. 2010). This study examined two nuclear genes, ANT and H3, from 376 individuals that were mostly the same as those examined in the previous mtDNA study.
PCR and sequencing
We screened ten candidate nuclear genes for this study. Initial methods for amplifications of ATP synthetase subunit α (ATPSα), ATP synthetase subunit β (ATPS β), adenine nucleotide transporter/ADP-ATP translocase (ANT), signal recognition particle 54-kDa subunit (SRP54), TATA box binding protein/transcription factor IID (TBP), lysidyl aminoacyl transfer RNA synthetase (LTRS) and zinc metalloproteinase (ZMP) were provided by Jarman et al. (2002). Only LTRS yielded clear sequences with Gemma but the fragments were not polymorphic. Primers for ANT, designed by Audijonyte and Vrijenhoek (2010), provided scorable polymorphic sequences, but their primers for calmodulin (CAL) and cyclophilin A (CYCA) could not be used with Gemma. New primers for H3 were designed based on Genbank sequence DQ184894.1 (Mikkelsen et al. 2006). Our criterion for selecting ANT and H3 in this study was that they reliably amplified and were polymorphic.
Nested polymerase chain reaction (PCR) methods were used to amplify ANT and H3 fragments. All PCR products were purified with a Multiscreen HTS PCR 96 vacuum manifold system (Millipore Corp. Billerica, MA), and then sequenced bi-directionally on an ABI3130 sequencer with BigDye Terminator v3.1 (Life Technologies Corp., Carlsbad, CA).
Initial amplifications of ANT used the primer pair
-
AntF_asta 5′ CCATTYTGGMGIGGWAACWTGGC3′ and
-
AntR1_asta 5′ TTCATCAAIGACATRAAICCYTC3′ (Audzijonyte and Vrijenhoek 2010)
Polymerase chain reaction protocol #1 consisted of 25 μL reactions with 1–2 μL DNA template, 15–20 pmol of each primer, 12 μL of the AmpliTaq Gold PCR reaction mix (Applied Biosystems Inc.), and 2 μL of 1 mg/mL Bovine Serum Albumin solution (BSA, New England Biolabs, Inc.). Touchdown PCR was applied following (Audzijonyte and Vrijenhoek 2010), with 94 °C for 10 min in the first cycle (1 min in subsequent cycles), annealing temperature starting at 55 °C for 1 min and decreasing 1 °C each cycle to 48 °C (7 cycle in totals), and 72 °C for 1 min, followed with 33 cycles of annealing temperature at 49 °C. Newly designed internal primers were used for the nested amplification:
-
AntF3_gm 5′ TAACGTGCTCAGGTACTTCC3′ and
-
AntR3_gm 5′AGATGTCATCATCATACGCCTT3′.
Nested amplifications involved Protocol #2, which consisted of 25 μL reactions with 1–2 μL DNA template, 12.5 μL AmpliTaq Gold® Fast PCR Master Mix (Applied Biosystems Inc.), 1 μL of each primer at 10 μmol/μL. PCR condition was 95 °C for 10 min, 35 cycles of 3 s denaturing at 96 °C, 3 s annealing at 56 °C, and 15 s extension at 68 °C, with a final extension at 72 °C for 7 min.
The new primers for initial amplifications of H3 are:
-
H3F1 5′TAGAAAATCCACCGGAGGWA3′ and
-
H3R1 5′ACGTTTGGCATGGATGGCGCAC3′
The nested amplifications we used the H3R1 in conjunction with an internal primer:
-
H3F2 5′AAGCTCCACGAAAGCAACTG3′.
Both the initial and nested amplifications used the PCR protocol #2 as previously listed.
Statistical analyses
Sequences were imported and aligned in Geneious 5.5 (Biomatters Ltd). Phase 2.1.1 (Stephens et al. 2001) was used to reconstruct allelic haplotypes from the ANT and H3 sequences. Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD) were evaluated in Genepop 4.0 (Raymond and Rousset 1995). The program CNDW (Asmussen and Basten 1994; Basten and Asmussen 1997) was used to test for cytonuclear disequilibrium in samples that included individuals assayed for mtDNA and the nDNAs. We used Arlequin 3.5 (Excoffier and Lisher 2010) to estimate haplotype diversity (h, Nei 1987), nucleotide diversity (π, Nei 1987), and Fu’s Fs (Fu and Li 1993). Allelic richness and private allelic richness were estimated using the rarefaction methods implemented in Hp-Rare 1.0 (Kalinowsky 2005).
For analyses of population structure, we generated multi-locus genotypes by recoding the nuclear sequences as alleles. These genotypes were concatenated with the previous COI sequences and examined in Structure 2.3 (Pritchard et al. 2000). Each analysis was conducted 20 times with K varying from 1 to 12. Each analysis involved 400 000 MCMC iterations after a burn-in period of 100,000. The optimum number of clusters was determined by comparing average log likelihood, Ln P(X|K), and delta K for each value of K (Evanno et al. 2005).
To estimate population sizes and immigration rates, we used the program IMa2 (Hey and Nielsen 2007), a coalescent-based method using Markov Chain Monte Carlo (MCMC) simulations. Our purpose was to compare demographic parameters between regions previously defined by the Structure analysis. Replicate runs were established with different prior estimates for θ, m, and t, with all yielding similar results. The maximum likelihood estimates of population sizes, and migration rates were left unscaled because the mutation rates for the nuclear genes were unknown.
Results
A 454 bp fragment of ANT was sequenced from 330 individuals. Haplotype reconstructions identified nine alleles. The numbers of alleles ranged from three to six in samples from the introduced population samples, and one to four in those from the native sites (Table 2). Only seven individuals were genotyped from the ME sample of 36 individuals. The remaining individuals failed to amplify after repeated attempts, due perhaps to a point mutation in one of the primer sites, because the quality of the DNA extracts was sufficient for amplifications of H3.
A 253 bp fragment of H3 was sequenced from 363 individuals. Haplotype reconstructions identified 10 alleles, which ranged from three to five in samples from the introduced sites and three to six alleles in samples from the native sites (Table 2).
Population diversity
For ANT, two population samples (BL, ES) showed significant heterozygote deficiencies, but a global test across populations was not significant (P > 0.05). In contrast, H3 exhibited significant excesses of heterozygotes in six of the 12 samples, and a global test remained significant for excess heterozygotes (P < 0.05). Nonetheless, the multi-locus test for departure from Hardy–Weinberg equilibrium of Raymond and Rousset (1995) obtained by summing across the two loci and all samples was not statistically significant. No significant linkage disequilibrium was observed between two nuclear loci (P > 0.05). Cytonuclear combinations involving the previous COI haplotypes also identified no evidence for gametic disequilibrium in any population sample.
Mean heterozygosities (H) and nucleotide diversities (π) for the two nuclear genes were compared between the East and West Coast population groups. The t tests of group means revealed no significant differences (P > 0.05) between the East and West Coast groups for either gene. No historical records indicated that the ME and NC were used as sources for Californian oyster stocks, so those samples were not included in group comparisons between native and introduced populations (This is also applied to other parameters below).
For ANT, the mean heterozygosities for the native and introduced populations were 0.38 and 0.47, respectively. The ANT*A allele was most frequent in a “northern” group of native samples (ME, MA, CT and NJ), whereas ANT*B was most frequent in a “Mid-Atlantic” group (MD and VA). ANT*C was relatively frequent in the Mid-Atlantic region and it dominated the southernmost sample from NC. This pattern was partially inverted in the introduced range, where ANT*A was most frequent in three “southern” samples (BL, SF and ES) and ANT*B was most frequent in two “northern” samples (TB and BH). ANT*C frequencies mirrored those found along the East Coast from MA to VA. Six singleton alleles were identified, with four occurring in the introduced population samples—BH (1), BL (1), SF (2)—and two occurring in a native site— NC (2).
For H3, mean heterozygosities for the native and introduced populations were 0.58 and 0.51, respectively. Two common haplotypes, H3*a and H3*b, occurred in all samples from both coasts. H3*d was frequent in the “Mid-Atlantic” and NC samples, and except for NJ, missing from the “northern” group of the native population. All of the introduced sampling sites, except BL, contained H3*d. One to two singletons were identified from some native or introduced population samples, except for TB, BL, ME, MA, and VA.
More singleton haplotypes were found in the previously reported COI data than were observed in both nuclear gene regions. Singletons had relatively higher frequency in the native population samples than in the introduced population (Fig. 1). The average percentage of singletons from the three markers are shown in suppl. Table 3, native samples had more singletons than the introduced sites, in general, but this was not significant (t test, P > 0.05)

Frequencies of ANT, H3, and COI haplotypes for Gemma gemma populations. Capital letters denote main ANT haplotypes, and small letters denote main H3 haplotypes. COI haplotypes from Hoos et al. (2010) after removing historical individuals. Uncolored pie slices represent singletons
Allelic richness (A r) and private allelic richness (P a) were estimated in Hp-Rare 1.0 (Kalinowsky 2005) after ME and NC being removed. The rarefaction included 32 genes per population sample, and 5 per region (Table 4). For ANT, similar values of A r were observed in native and introduced populations, with west coast showing higher overall A r than that of east coast (4.53 vs. 3.00). P a showed a similar pattern but this was mostly driven by the high values from BL and SF (Table 4). For H3, both allelic richness and private allelic richness were relatively higher in native than in introduced samples. This is also true for the regional comparison. When we analyzed the two nuclear markers together, the east region showed higher values than that of west region both in A r (5.07 vs. 4.37) and P a (1.57 vs. 0.87). We also estimated the allelic richness and private allelic richness based on previous COI data, and those from the native region were roughly twofold higher than that of the introduced population. Especially high allelic richness and private allelic richness were found in ES and CT.
Population subdivision
Allelic data for the 12 samples were analyzed with the program Structure 2.3 which identified two population clusters (Fig. 2). An analysis of DeltaK values rejected all clusters greater than K = 2. The average log likelihood, Ln P(X|K), was lowest for K = 1; thus, we accepted a two-cluster model as the best description of the nuclear genotypic data. Inclusion of the mitochondrial COI haplotypes in this analysis strengthened the conclusion that K = 2 provided the best model for these samples (Fig. 2b). The same two clusters appeared on both the east and west coasts, but the latitudinal order of these groupings was inverted on the west coast. As previously noted, this inversion was apparent in the raw allelic frequencies (Fig. 1). The northernmost samples on west coast (BH and TB) clustered together with three southern samples (MD, VA and NC) from the east coast, and the three southern sampling areas on the west coast (BL, SF and ES) clustered with northern ones from the east coast.

Bar plots from STRUCTURE analysis at K = 2 with a or without b COI. Populations from west coast presented on left. Each individual genotype is represented as a vertical bar with different shades representing the posterior probabilities of membership in K inferred clusters
Taken alone, the east coast populations exhibited a distinct phylogeographic break between NJ and MD. Analysis of the nuclear and mitochondrial sequences with the Isolation-with-Migration program, IMa2, revealed demographic differences between the northern and southern clusters (Fig. 3, Table 5). Estimated population sizes (θ’s) were broadly overlapping between two regions (means 1.100 vs. 1.486). Ancestral population size, θA, was not estimated because of the flat posterior probability distribution (Fig. 3b). Based on the historical evidence for introductions of southern oyster stocks into the northern regions, we predict that southern stowaways also contributed to a northward migration of Gemma. As expected, the maximum likelihood estimate for immigration of southern alleles into the northern population cluster (m N>S = 3.683) was greater than that for the reverse direction (m S>N = 1.244); nonetheless the HPD intervals overlapped (Table 5).

Multilocus estimates from IMA2 of demographic parameters from east coast populations, ME and NC excluded. The 95 % highest and lowest posterior density (HPD) intervals are shown
Discussion
Loss of genetic diversity
Population bottlenecks are predicted to reduce allelic richness (A r) more rapidly than heterozygosity (H), because rare alleles are more easily lost by chance than are high frequency alleles (Nei 1987). This study identified no significant differences in the heterozygosity of nuclear DNA (nDNA) sequences from two genes of native and introduced populations of the amethyst gem clam, Gemma gemma. The results are consistent with a previous analysis of haplotypic diversities (h) for mitochondrial DNA COI (Hoos et al. 2010). However as expected, estimates of allelic richness were greater in the native source populations. The overall percentage of singleton alleles (P), a useful index of population bottlenecks (Neilson and Wilson 2005), was low in some California population samples (e.g. TB and BL). Curiously, P was not greatest in San Francisco Bay (SF) samples, the site of repeated eastern oyster introductions. The highest P values occurred in Elkhorn Slough (ES) samples, a site that received repeated secondary oyster explants from San Francisco Bay. Why this secondary location retained more diversity than the primary site of transplants is unknown; however, Hoos et al. (2010) suggested that Elkhorn Slough may have had additional sources from the Gulf of Mexico, if historical records of Gemma there are correct. Both heterozygosity and allelic richness were low in the northernmost Atlantic Coast sample of Gemma from Maine (ME). Overexploited native populations in this region likely received fewer explants from the more diverse Mid-Atlantic region. The southernmost sample from the native populations (NC) was relatively diverse in both the nuclear and mitochondrial DNA regions. Gene flow from southerly Gemma populations may be responsible for this effect, but this hypothesis remains to be tested with the acquisition of additional samples from the southeastern US and potentially from the Gulf Coast region.
Population subdivision in native populations
A phylogeographic break between native regions previously identified by COI (Hoos et al. 2010) also was confirmed with the present nDNA sequences. Multilocus Structure analyses clearly identified “northern” and “Mid-Atlantic” population clusters, with and without inclusion of the mtDNA sequences. Concordant phylogeographic patterns have been observed in a number of US East Coast marine taxa, for example, the killifish Fundulus heteroclitus (Adams et al. 2006), the polychaete Marenzellaria viridis (Bastrop et al. 1998), the sea stars Asterias rubens/forbesi (Wares 2001), and the mussel Geukensia demissa (Diaz-Ferguson et al. 2010). The region between Upper and Lower Virginian provinces appears to present a common contact zone for estuarine species (Diaz-Ferguson et al. 2010). Nonetheless, historical processes responsible to generating these concordant patterns in multiple taxa, are not clearly resolved (reviewed in Wares 2002). Historical isolation of taxa in “northern” refugia during glacial maxima followed by secondary contact might have created the overall patterns. Nonetheless, other factors might be required to stabilize the contact zones in taxa with high rates of dispersal. For example, physical barriers to dispersal such as ocean currents, strong selection for physiological races, habitat selection, and incipient reproduction isolation might be involved (reviewed in Avise 2000).
We can revisit several of these hypotheses with respect to Gemma. The history of oyster transplants may shed some light on the mechanisms responsible for population subdivision along the US East Coast. Historically, eastern oysters from Chesapeake Bay were dredged and transported to Delaware, New Jersey, New York and the New England states (Ingersoll 1881). Approximately 60 % of oysters transported to New England through New Jersey originated from Chesapeake Bay in 1880 (Miller 2000). If Gemma stowaways were simultaneously transported, we should have observed historical evidence for northward gene flow. Alleles that arose in Mid-Atlantic populations should have introgressed into the “northern” populations. Structure analysis involving the two nuclear genes provided some evidence for admixture in the NJ sample, but not for more northern populations. Inclusion of mitochondrial COI in the Structure analysis clearly identified two population clusters with limited evidence for immigration between Mid-Atlantic and the “northern” clusters. Although stronger northward gene flow was identified by IMa2, the known history of oyster transplants did not correspond to the pattern of population subdivision in Gemma. Invoking a dispersal barrier for native Gemma populations seems unlikely in this case; however, regional selection to maintain life history differences or physiological adaptations might significantly limit the gene flow between discrete geographical races of a species (Pelc et al. 2009). Consequently, habitat selection may have played a significant role in maintaining the difference between East Coast populations of Gemma.
As previous noted, the north–south pattern of subdivision appeared inverted in the introduced Gemma populations. A simple hypothesis for the this ‘antiparallel’ pattern invokes separate primary introductions (Hoos et al. 2010), one from Mid-Atlantic region to Tomales Bay and Bodega Harbor, and another from the Northeastern region to San Francisco Bay, Bolinas Lagoon and Elkhorn Slough. Nonetheless, no historical records exist for direct introductions of Mid-Atlantic oyster stocks to Tomales Bay and Bodega Harbor. Large numbers of Eastern oysters (and presumably Gemma stowaways) derived from the New York harbor region (Barrett 1963), which were mixed with Mid-Atlantic explants. Historical records indicate that these mixed stocks provided the transplants to San Francisco Bay. We are aware of no evidence that Tomales Bay and Bodega Harbor are environmentally more similar to the Mid-Atlantic environments.
References
Adams SM, Lindmeier JB, Duvernell DD (2006) Microsatellite analysis of the phylogeography, Pleistocene history and secondary contact hypotheses for the killifish, Fundulus heteroclitus. Mol Ecol 15:1109–1123
Allendorf FW (1986) Genetic drift and the loss of alleles versus heterozygosity. Zoo Biol 5:181–190
Allendorf FW, Lundquist LL (2003) Introduction: population biology, evolution, and control of invasive species. Conserv Biol 17:24–30
Asmussen MA, Basten CJ (1994) Sampling theory for cytonuclear disequilibria. Genetics 138:1351–1363
Audzijonyte A, Vrijenhoek RC (2010) Three nuclear genes for phylogenetic, SNP and population genetic studies of molluscs and other invertebrates. Mol Ecol Resour 10:200–204
Avise JC (2000) Phylogeography: the history and formation of species. Harvard University Press, Cambridge
Bachtrog D, Thornton K, Clark A, Andolfatto P (2006) Extensive introgression of mitochondrial DNA relative to nuclear genes in the Drosophila yakuba species group. Evolution 60:292–302
Barrett EM (1963) The California oyster industry. Calif Dept Fish Game Fish Bull 123:1–103
Basten CJ, Asmussen MA (1997) The exact test for cytonuclear disequilibria. Genetics 146:1165–1171
Bastrop R, Jurss K, Sturmbauer C (1998) Cryptic species in marine polychaete and their independent introduction from North America to Europe. Mol Biol Evol 15:97–103
Behar DM, Hammer MF, Garrigan D, Villems R, Bonne-Tamir B, Richards M, Gurwitz D, Rosengarten D, Kaplan M, Della Pergola S, Quintana-Murci L, Skorecki K (2004) MtDNA evidence for a genetic bottleneck in the early history of the Ashkenazi Jewish population. Eur J Hum Genet 12:355–364
Boratyński Z, Alves PC, Berto S, Koskela E, Mappes T, Melo-Ferreira J (2011) Introgression of mitochondrial DNA among Myodes voles: consequences for energetics? BMC Evol Biol 11:355
Brown JE, Stepien CA (2010) Population genetic history of the dreissenid mussel invasions expansion patterns across North America. Biol Invasions 12(11):3687–3710
Carlton JT (1979) History, biogeography, and ecology of the introduced marine and estuarine invertebrates of the Pacific Coast of North America. Ph.D. Dissertation, University of California, Davis, CA
Cornuet J, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Diaz-Ferguson E, Robinson JD, Silliman B, Wares JP (2010) Comparative phylogeography of North American Atlantic salt marsh communities. Estuaries Coasts 33:828–839
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lisher HEL (2010) Arlequin ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Fu YX, Li W-H (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Galtier N, Nabholz B, Glemin S, Hurst GDD (2009) Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol Ecol 18:4541–4550
Gillis NK, Walters LJ, Fernandes FC, Hoffman EA (2009) Higher genetic diversity in introduced than in native populations of the mussel Mytella charruana: evidence of population admixture at introduction sites. Divers Distrib 15:784–795
Hey J, Nielsen R (2007) Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc Natl Acad Sci USA 104:2785–2790
Hoos P, Geller J, Ruiz G, Miller W, Vrijenhoek RC (2010) The introduction of western Atlantic Gemma gemma to California: comparing inferences from genetic and historical data. Biol Invasions 16:582–592
Ingersoll EA (1881) The oyster-industry of the United States. In: Goode GB (ed) The history and present condition of the fishery industries, 1879–1889. Government Printing Office, Washington, DC
Jarman SN, Ward RD, Elliott NG (2002) Oligonucleotide primers for PCR amplification of coelomate Introns. Mar Biotechnol 4:347–355
Kalinowsky S (2005) HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189
Kochiss JM (1974) Oystering from New York to Boston. Mystic Seaport Inc, Middletown
Kolbe JJ, Glor RE, Schettino LRG, Lara AC, Larson A, Losos JB (2004) Genetic variation increases during biological invasion by a Cuban lizard. Nature 431:177–181
Mikkelsen PM, Bieler R, Kappner I, Rawlings TA (2006) Phylogeny of Veneroidea (Mollusca: Bivalvia) based on morphology and molecules. Zool J Linn Soc 148:439–521
Miller AW (2000) Assessing the importance of biological attributes for invasion success: eastern oyster (Crassostrea virginica) introductions and associated molluscan invasions of the Pacific and Altantic coastal system. Ph.D. Dissertation, University of California, Los Angeles
Nei M (1975) Molecular population genetics and evolution. North-Holland Pub. Co., Amsterdam
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution 29:1–10
Neilson ME, Wilson RR (2005) mtDNA singletons as evidence of a post-invasion genetic bottleneck in yellowfin goby Avanthogobius flavimanus from San Francisco Bay, California. Mar Ecol Prog Ser 296:197–208
Pelc RA, Warner RR, Gaines SD (2009) Geographical patterns of genetic structure in marine species with contrasting life histories. J Biogeogr 36:1881–1890
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raymond M, Rousset F (1995) GENEPOP (Ver. 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Roman J, Darling JA (2007) Paradox lost: genetic diversity and the success of aquatic invasions. Trends Ecol Evol 22:454–464
Ruiz GM, Fofonoff PW, Ashton G, Minton MS, Miller AW (2013) Geographic variation in marine invasions among large estuaries: effects of ships and time. Ecol Appl 23:311–320
Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989
Wares JP (2001) Biogeography of Asterias: North Atlantic climate change and speciation. Biol Bull 201:95–103
Wares JP (2002) Community genetics in the Northwestern Atlantic intertidal. Mol Ecol 11:1131–1144
Acknowledgments
The authors thank Phillip M. Hoos and Whitman Miller for sample collection and DNA extraction, and Julio Harvey, Shannon Johnson and Kristine Walz for helpful comments and sequencing assistance. We thank Associate Editor Carol Stepien for her careful editorial assistance. This research was supported by the Monterey Bay Aquarium Research Institute with funds from The David and Lucile Packard Foundation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, H., Geller, J.B. & Vrijenhoek, R.C. Genetic diversity in native and introduced populations of the amethyst gem clam Gemma gemma (Totten, 1834) from the U.S. east and west coasts. Biol Invasions 16, 2725–2735 (2014). https://doi.org/10.1007/s10530-014-0699-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-014-0699-9