
Abstract
In this study, we isolated and characterized an insect nidovirus from the mosquito Culex tritaeniorhynchus Giles (Diptera: Culicidae) in Vietnam, as an additional member of the new family Mesoniviridae in the order Nidovirales. The virus, designated “Dak Nong virus (DKNV),” shared many characteristics with Cavally virus and Nam Dinh virus, which have also been discovered recently in mosquitoes, and these viruses should be considered members of a single virus species, Alphamesonivirus 1. DKNV grew in cultured mosquito cells but could not replicate in the cultured vertebrate cells tested. N-terminal sequencing of the DKNV structural proteins revealed two posttranslational cleavage sites in the spike glycoprotein precursor. DKNV is assumed to be a new member of the species Alphamesonivirus 1, and the current study provides further understanding of viruses belonging to the new family Mesoniviridae.
Similar content being viewed by others

Introduction
The members of the order Nidovirales are enveloped viruses that have a linear, single-stranded RNA genome of positive polarity with a 5′ cap structure and a 3′ poly (A) tail [1]. This virus order currently comprises four families that include nine genera [2]: Arteriviridae (genus Arterivirus), Coronaviridae (divided into two subfamilies, Coronavirinae [genera Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus) and Torovirinae (genera Torovirus and Bafinivirus]), Roniviridae (genus Okavirus), and Mesoniviridae (genus Alphamesonivirus). The Ninth Report of the International Committee on Taxonomy of Viruses lists approximately 80 nidoviruses, many of which infect birds and mammals, including humans, livestock, and companion animals [1]. In contrast, bafiniviruses have been isolated from freshwater fishes [3, 4], and roniviruses are the only invertebrate nidoviruses known to infect prawns. Coronaviruses and roniviruses, which have genomes larger than 26 kb, are categorized as “large nidoviruses”; they are the largest RNA viruses known to date. In contrast, arteriviruses are referred to as “small nidoviruses” because of their 13-16 kb genome size.
Despite considerable differences in genome size and gene composition, all nidoviruses have similar genome organizations and replication strategies [1]. The large 5′ part of the genome encodes two partially overlapping ORFs, which are generally designated ORF1a and 1b. Translation of ORF1a yields a polyprotein called pp1a. ORF1b is translated as a fusion with the ORF1a product, to form polyprotein pp1ab, by a programmed −1 ribosomal frameshift (RFS) at the overlap site, which contains a specific slippery sequence just upstream of a pseudoknot structure. The large polyproteins pp1a and pp1ab are proteolytically processed to yield the replicase subunits, including a 3C-like (3CL) protease flanked by transmembrane (TM) domains, which is encoded by ORF1a, and both an RNA-dependent RNA polymerase (RdRp) and a superfamily 1 helicase (HEL1) encoded by ORF1b [5]. The region downstream of ORF1b contains multiple smaller ORFs, whose number may vary among viruses, which encode a set of viral structural proteins (e.g., nucleocapsid protein and spike glycoprotein). Translation of these structural proteins occurs through a nested set of 3′-coterminal subgenomic mRNAs that are controlled by leader transcription-regulating sequences (TRSs) and have different 5’ ends depending on the nidovirus family and genus [6].
Recently, two new nidoviruses have been discovered in mosquitoes in different regions. A short genome fragment of Cavally virus (CavV), the first nidovirus isolated from mosquitoes in Côte d’Ivoire, was reported in 2009 [7], and its detailed characterization was reported in 2011 [8]. Soon after that, isolation and characterization of Nam Dinh virus (NDiV) from mosquitoes in Vietnam was reported [9]. The two new viruses have genome organizations and gene expression patterns that are essentially similar to those of known nidoviruses, and it is noteworthy that their genome sizes (approximately 20 kb) are intermediate between those of large and small nidoviruses. Subsequent detailed analysis of the CavV and NDiV genomes implied that they should be considered members of a single virus species, Alphamesonivirus 1, which has been assigned to a new family, Mesoniviridae (genus Alphamesonivirus), in the order Nidovirales [10]. As these viruses were only recently discovered, their ecological aspects (e.g., host range, life cycle, and pathogenicity) remain to be elucidated.
In this study, we describe the isolation and characterization of a virus from the mosquito Culex tritaeniorhynchus Giles (Diptera: Culicidae) in Vietnam. The virus, designated “Dak Nong virus (DKNV),” has characteristics similar to CavV and NDiV in virion morphology, genomic sequence, and genome organization. Furthermore, we present the growth properties of DKNV in several cultured cell lines and describe the processing of the spike glycoprotein precursor, which provides further understanding of viruses belonging to the new family Mesoniviridae.
Materials and methods
Mosquito collection
Female mosquitoes of the species C. tritaeniorhynchus were captured in Dak Nong Province in the highlands of Vietnam in July 2007 [11]. Collected mosquitoes were sorted into pools and kept frozen at −80 °C.
Cell culture and virus isolation
The mosquito cell line C6/36 (HSRRB, Osaka, Japan) was used for further study. Cell culture and virus isolation were done as described previously [12]. In brief, supernatants of mosquito homogenates were passed through sterile 0.45-μm filters. The filtrates were diluted and inoculated onto monolayers of C6/36 cells, and the cells were incubated for 7 days. After two additional blind passages, the supernatants were harvested and stored as viral stocks at −80 °C.
Electron microscopic analysis
The culture supernatant from infected C6/36 cells showing CPE was centrifuged at 1,600 × g for 15 min, and the supernatant was centrifuged again at 12,000 × g for 30 min to remove cellular debris. The fluid was fixed in 2 % glutaraldehyde and processed for negative-stain electron microscopy with 2 % phosphotungstic acid.
Plaque purification and titration of the virus
Monolayer cultures of C6/36 cells grown in 6-well culture plates were inoculated with tenfold serial dilutions of the virus stocks. After 1 h of virus adsorption, cells were covered with overlay medium (MEM containing 1 % SeaPlaque GTG Agarose [TAKARA BIO, Shiga, Japan], 2 % FBS, and 2 % non-essential amino acids). Two days later, a single plaque was collected and suspended in the culture medium, which was inoculated onto C6/36 cells to propagate the plaque-purified strain, HL30.
Four mammalian cell lines, Vero (African green monkey kidney), BHK-21 (baby Syrian hamster kidney), HmLu-1 (hamster lung) and CCL-141 (duck embryo), and three mosquito cell lines, C6/36, NIAS-AeAl-2 (Aedes albopictus) [13], and NIID-CTR (C. tritaeniorhynchus) [14], were used to examine the host range of the HL30 strain. Each cell line (2 × 106 cells) was plated in a 25-cm2 culture flask and inoculated with virus at an m.o.i. of 0.01 PFU/cell. Two hours after infection, the inoculum was removed and replaced by fresh medium. Small aliquots of the medium were recovered at periodic intervals, and each aliquot was titered by plaque assay on C6/36 cells [15, 16].
Nucleotide sequence determination and analysis of the viral genome
To identify the infectious agent HL30, we analyzed HL30-infected cell cultures using the rapid determination of viral RNA sequences (RDV) system, a method for sequence-independent amplification of viral genome sequences [17]. After obtaining the infection-specific sequences by the RDV system, specific internal primers were designed to amplify unknown flanking regions and to fill gaps in the viral genome sequence. First-strand cDNA was generated using a SuperScript III Reverse Transcription System (Invitrogen, Carlsbad, CA, USA). PCR was performed using KOD Plus-ver. 2 (Toyobo Co. Ltd., Tokyo, Japan). To determine the 5′- and 3′-terminal sequences of the viral genome, RACE was performed using a GeneRacer Kit (Invitrogen).
RNA secondary structures were predicted using the DotKnot program [18, 19]. The deduced amino acid (aa) sequences of each ORF were analyzed for predictions of hydrophilicity using TMpred [20], transmembrane helices using TMHMM 2.0 [21], and signal peptides using SignalP 4.0 [22]. Prediction of the cleavage sites in the putative spike glycoprotein precursor was performed using ProP 1.0 [23], and coiled-coil structure prediction analysis of the precursor was conducted using the COILS [24] and Paircoil 2 [25] servers in the default configuration. Prediction of protein binding and order/disorder regions was conducted using the ANCHOR server [26].
Identification of transcription-regulating sequences for subgenomic mRNAs
To identify TRSs for DKNV, 5′-terminal sequences of viral subgenomic mRNAs were determined using 5′-RACE analysis. In brief, total RNAs were extracted from infected C6/36 cells using an RNeasy Mini Kit (QIAGEN Inc., Venlo, The Netherlands). Poly(A)+ RNAs were separated from total RNA using a Poly(A)+ Isolation Kit from Total RNA (Nippon Gene Co., Tokyo, Japan) and then were subjected to 5′-RACE analysis using a GeneRacer Kit (Invitrogen).
Northern hybridization
Viral transcripts were examined by northern hybridization using DIG-labeled antisense RNA probes. A 481-bp region (sequence position 19740–20220) was selected for the probes, which were initially amplified by RT-PCR. The PCR product containing a complete T7 promoter sequence was used as the template for generating DIG-labeled RNA probes with a DIG RNA Labeling Kit (Roche Diagnostics, Tokyo, Japan). Total RNA extracted from infected C6/36 cells was electrophoresed in a 0.8 % denaturing formaldehyde agarose gel, transferred to a nylon membrane (Hybond-N+; GE Healthcare, Uppsala, Sweden), and then UV cross-linked to the membrane. After a prehybridization, the probe was hybridized to the blot in DIG Easy Hyb (Roche Diagnostics) at 68 °C overnight. The membrane was washed twice with 2×SSC (0.1 % SDS) at RT for 10 min and 0.1×SSC (0.1 % SDS) at 68 °C for 20 min. The RNA-RNA hybrid was detected using immunodetection with alkaline phosphatase-conjugated anti-DIG antibody Fab fragments in blocking solution, and the hybridization signals were captured using CDP-Star (Roche Diagnostics).
Analysis of viral structural proteins
One hundred milliliters of supernatant taken from C6/36 cell cultures inoculated with HL30 and incubated for 2 days were used for virus purification. The fluid was clarified from cell debris by low-speed centrifugation and mixed with polyethylene glycol (PEG) solution (final concentration, 8 % PEG 6000, 0.5 M NaCl) at 4 °C overnight. The fluid was centrifuged (4500 × g, 30 min, 4 °C), and the precipitate was resuspended in Dulbecco’s PBS (−) (DPBS; Gibco BRL, Gaitherburg, MD, USA). The suspension was layered on 3 ml of a linear sucrose gradient (20–70 % in DPBS) and ultracentrifuged (85,000 × g, 16 h, 4 °C). The fraction containing the virus band, visualized by illumination, was collected, dialyzed against DPBS to remove the sucrose, and analyzed for viral structural proteins. Protein samples for SDS-PAGE analysis were prepared by mixing the purified virus suspension with SDS sample buffer in the presence of 2-mercaptoethanol and then heating it at 95 °C for 3 min. The viral proteins were electrophoresed on a 15–20 % gradient tricine polyacrylamide gel (Wako Pure Chemical Industries) and stained using Oriole Fluorescent Gel Stain (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
The N-terminal sequences of representative proteins were determined by Edman degradation. Viral protein bands on a gel were transferred to a PVDF membrane (Bio-Rad Laboratories) in transfer buffer using a semi-dry blotter (Bio Craft, Co., Ltd., Tokyo, Japan). The protein bands stained with SimplyBlue Safestain (Life technologies, Carlsbad, CA, USA) were excised from the membrane and subjected to N-terminal sequencing using an Applied Biosystems 494/HT PROCISE Sequencing System.
Phylogenetic study
Bayesian phylogenetic inference, based on the protein sequences of the core motif in the RdRp domains, was conducted using selected members of the order Nidovirales. The inferred aa sequences of the F3 domain as well as the A, B, and C domains of DKNV were aligned together with the corresponding regions of 19 nidoviruses [27, 28]. A tree was constructed using the program Mrbayes 3.1.2 [29] for mrmodeltest analysis with a WAG substitution matrix [30]. In addition, the aligned matrix data were also analyzed using neighbor-joining (NJ) and maximum-likelihood (ML) algorithms with the JTT models [31] using the software package PHYLIP ver. 3.69 [32] to avoid algorithm bias. The statistical significance of the resulting trees was evaluated using a bootstrap test with 1,000 replications [33]. All trees were represented graphically using TreeView software ver. 1.6.6 [34].
Nucleotide sequence accession number
The complete genome sequence of DKNV has been deposited in the GenBank/EMBL/DDBJ database under accession number AB753015.
Results
Isolation, electron microscopy, and growth kinetics of the new virus
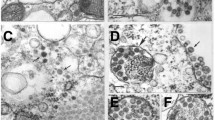
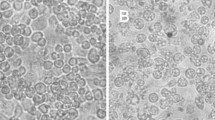
During the course of arbovirus surveillance in mosquitoes from Vietnam [11], we observed that two pools of mosquito homogenate from 131 pools (4,199 mosquitoes) of C. tritaeniorhynchus induced severe CPE in C6/36 cultured mosquito cells. Cell growth was significantly suppressed by the agents, and almost all cells were detached from the surface of the culture plate within 48-h postinfection (Fig. 1). Electron microscopic observation revealed that enveloped, spherical virus-like particles with an average diameter of approximately 80 nm were present in the infected cell culture supernatant (Fig. 2a). Surface projections that protrude from the envelope were observed on the particles (Fig. 2b).

(a) Phase-contrast micrographs of mock-infected (left) and DKNV-infected (right) C6/36 cells at 2 days postinfection. Scale bars indicate 100 μm. (b) Plaque morphology induced by DKNV infection of a C6/36 cell monolayer in a 6-well plate (Corning Inc., NY, USA)

Negative-contrast electron micrographs of DKNV particles
The virus was plaque purified on C6/36 cells, and a purified strain designed HL30 induced severe CPE in C6/36 cells similar to the unpurified original isolate. HL30 could replicate in two other mosquito cell lines but not in the cultured vertebrate cell lines tested (Fig. 3). Interestingly, HL30 grew to high titers (approximately maximum titer 1011 PFU/mL within 36 h post-infection) in two A. albopictus cell lines, C6/36 and NIAS-AeAl-2, but grew poorly (approximate maximum titer, 102 PFU/mL) in the C. tritaeniorhynchus cell line NIID-CTR.

Growth kinetics of DKNV in four vertebrate cell lines (Vero, BHK-21, HmLu-1 and CCL-141) and three mosquito cell lines (C6/36, NIAS-AeAl-2, and NIID-CTR)
Genetic characterization of the new virus
We applied the RDV system [17] to identify the isolated viruses. The PCR products from the final step of the RDV system were sequenced and compared to the public sequence databases. Several PCR amplicons that proved to be infection-specific had significant homology to the nucleotide sequences of two recently discovered insect nidoviruses, CavV [8] and NDiV [9], which have been recently assigned to be the same virus species, Alphamesonivirus 1 [10]. Based on virion morphology and the sequence homology, we concluded that the infectious agent could be an insect nidovirus related to CavV and NDiV, and we named it as “Dak Nong virus (DKNV)”, after the province of the mosquito collection site in Vietnam.
The complete genome sequence of DKNV (strain HL30) was 20256 bp in length excluding the 3′ poly (A) tail. The genome organization closely resembles that of the nidoviruses [1], particularly viruses of the species Alphamesonivirus 1 [10]. The DKNV genome appeared to contain at least seven ORFs, ORF1a (bp 371–7984), ORF1b (bp 8245–15744), ORF2a (bp 15768–18494), ORF2b (bp 15782–16450), ORF3a (bp 18538–19011), ORF3b (bp 18887–19237), and ORF4 (bp 19547–19684) (Fig. 4a). The coding region of the genome is flanked by a 5′- untranslated region (UTR) (bp 1–370) and a 3′-UTR (bp 19685–20256) followed by the poly (A) tail.

Genome organization and gene expression of DKNV. (a) Schematic diagram of the organization of the DKNV genome. Horizontal lines and rectangles indicate the viral genomic RNA and the locations of ORFs arranged in three reading frames (−1, 0, +1), respectively, relative to that of ORF1a. Putative protein domains encoded by ORF1a/b are highlighted in gray. The predicted site of RFS is indicated by a black dot. (b) Schematic representation of DKNV gene expression. Horizontal lines and open boxes on the lines indicate the DKNV mRNAs (mRNA1–4) and ORFs corresponding to the positions shown in panel a, respectively. Two different leader TRSs that regulate the expression of DKNV subgenomic mRNA2 and 3 are shown in a gray-dotted box. Northern hybridization analysis (bottom left) identified at least two bands that correspond to the predicted sizes of subgenomic mRNA2 (upper) and mRNA3 (lower)
Like the current members of the species Alphamesonivirus 1, the two largest ORFs of DKNV contained several conserved domains found in all nidoviruses: a 3CL protease domain flanked by two TM domains that is encoded within ORF1a, and both an RdRp domain and a HEL1 domain that are encoded within ORF1b (Fig. 4a). Two other replicative domains common to nidoviruses (except arteriviruses), a 3′-to-5′ exoribonuclease (EXON) and a ribose-2′-O-methyltransferase (O-MT) were also identified in the C-terminal portion of DKNV ORF1b. In addition, like roniviruses and viruses belonging to Alphamesonivirus 1 (CavV and NDiV) [10], a putative N7-methyltransferase (N-MT) was also identified in the region between the EXON and O-MT domains, but not the uridylate-specific endoribonuclease (NendoU) domain that is commonly found in most nidoviruses [35–37].
The DKNV ORF2a, which is expected to encode a spike glycoprotein, encodes a 908-aa protein that contains five stretches of hydrophobic residues (Fig. S1); it has a predicted molecular mass of 103.8 kDa (Table 1). The program SignalP 4.0 predicted that signal peptidase-mediated cleavage could occur just after the second hydrophobic stretch (aa position 231C↓S) and that the C-terminal hydrophobic stretch could be a TM domain (aa position 889–908). DKNV ORF2b, which encodes a putative nucleocapsid protein, encodes a 222-aa (24.7 kDa) protein containing 12 proline (5 %), 34 basic (15 %), 18 acidic (8 %) and no cysteine residues. This protein was predicted to be highly basic (pI = 10.26) and hydrophilic (Fig. S1). The DKNV ORF3a and ORF3b encodes a 157-aa and a 116-aa protein with predicted molecular masses of 17.4 kDa (pI = 8.13) and 13.9 kDa (pI = 9.3), respectively. The deduced aa sequence of the ORF3a product was predicted to contain an N-terminal signal sequence (aa position 1–17) and a single-pass TM domain (aa position 94–116). In contrast, the putative protein encoded by ORF3b possessed an N-terminal hydrophilic region and a C-terminal hydrophobic region predicted to have a double-spanning TM topology (Fig. S1); such an amphiphilic nature is typical for matrix proteins of nidoviruses [8].
Analysis of viral transcripts
To identify the TRSs of DKNV [6], the 5′-terminal sequences of viral subgenomic mRNAs were analyzed by 5′-RACE using gene-specific primers derived from the sequences of ORFs 2, 3, and 4. The leader TRSs for generating subgenomic mRNA2 (for expressing ORF2a and 2b proteins) and mRNA3 (for expressing ORF3a and 3b proteins) were identified at sequence regions 124–144 and 26–40 of the viral genome, respectively, indicating that at least two different leader TRSs were utilized to generate subgenomic mRNAs (Fig. 4b). The 5′-terminal region of putative mRNA4 (for expressing ORF4 proteins) was not amplified in this experiment.
To confirm the presence of DKNV subgenomic mRNAs, northern blotting was performed with a DIG-labeled RNA probe specific for the 3′-terminal region of the DKNV genome. Two strongly hybridizing bands were detected at approximately 4.5 kbp and 1.8 kbp in size (Fig. 4b), which were almost identical to the expected sizes of mRNA2 and mRNA3, respectively. Like CavV [8], a weakly hybridizing band of uncertain origin was also detected at approximately 2.7 kbp in size (Fig. 4b); further studies are needed to identify this RNA species. Unlike CavV [8] and NDiV [9], no large RNA species (more than 10 kbp) derived from DKNV mRNA1 (for expressing ORF1a and ORF1ab proteins) was detected in this study, which may because of the difference in detection methods. We were also unable to obtain evidence for the expression of DKNV mRNA4 by northern blotting. This result is consistent with the 5′-RACE analysis described above, indicating that ORF4 expression in mesoniviruses needs further validation.
Prediction of the RNA secondary structure around the ORF1a/b overlapping site
As has been observed in NDiV [9], the overlap region between DKNV ORF1a and ORF1b contains a putative ‘slippery sequence (GGAUUUU)’ required for −1 ribosomal frameshifting (Fig. 5a). Accordingly, DKNV ORF1b could only be translated as a fusion with the ORF1a product, resulting in the generation of two forms of replicase polyprotein, pp1a and pp1ab. An RNA secondary structure analysis predicted a pseudoknot structure located immediately downstream of the slippery sequence (Fig. 5b).

Predicted RFS element and RNA prseudoknot structure in the DKNV genome. (a) The genomic nucleotide sequences (upper) and deduced amino acid sequences (lower) of ORF1a and ORF1b around their overlapping site. The deduced amino acid sequence of DKNV pp1ab is shaded. (b) A putative RNA pseudoknot structure immediately downstream of a “slippery sequence (GGAUUUU)” predicted using the DotKnot program [18, 19]
Analysis of viral structural proteins
SDS-PAGE analysis showed that the DKNV virion was composed of at least four major polypeptides with apparent molecular masses of approximately 80, 55, 30, and 20 kDa and several minor polypeptides with apparent molecular masses of approximately 29, 28, and 16 kDa (tentatively named p80, p55, p30, p20 for major bands, and p29, p28, and p16 for minor bands, respectively) (Fig. 6a). As is the case for NDiV [9], p30, p20, and p16 could be the protein products encoded by ORF2b, ORF3a, and ORF3b, respectively. The differences in relative protein amounts between p20 and p16 might be reflected by the translation efficiencies of ORF3a and ORF3b attributed to the position of each start codon on mRNA3. The apparent molecular mass of the ORF2b product (p30) was slightly larger than the mass calculated from the aa sequence data (25 kDa), probably due to the slow mobility on SDS gels attributable to its highly polar arrangement of charged residues [38, 39].

(a) SDS-PAGE and N-terminal sequencing analyses of DKNV virion proteins. *The determined N-terminal aa sequences of p80 and p55 are shown in arrowhead-shaped boxes. **The N terminus of p30 could not be directly sequenced because of the presence of a blocked N-terminal residue. (b) Cleavage sites in the precursor ORF2a polypeptide. An arrow and an arrowhead represent the cleavage sites of signal peptidase and furin-like proteases, respectively. (c) Predicted topology of the two forms of DKNV spike glycoprotein, msS1 and msS2. Cylinders and solid lines represent predicted transmembrane domains and ectodomains, respectively. The precursor ORF2a polypeptide is posttranslationally processed by signal peptidase to yield msS1 and further processed by a furin-like protease to yield msS2
In general, the spike glycoproteins of nidoviruses undergo posttranslational processing during virion morphogenesis [1]. The spike glycoprotein of NDiV, which has been shown to be encoded by ORF2a, migrates as a single prominent band on SDS gels with a molecular mass of 77 kDa [9]. In DKNV, two different protein bands (p80 and p58) seemed to be proteolytically processed forms of the ORF2a product. The ProP program predicted a furin-type cleavage site (prediction score +0.64 at position 209SQKRTKR↓WD) within DKNV ORF2a, which could be involved in post-translational processing of the precursor protein. To identify these two peptides and verify the putative cleavage site, N-terminal amino acid sequence analyses were conducted on three representative protein bands: p80, p55, and p30. The p80 and p58 peptides had different N-terminal sequences, both of which were encoded within the ORF2a sequence (Fig. 6a), indicating that these proteins could be generated through post-translational cleavages of a full-length ORF2a product. As pointed out before, the first cleavage site was located just after a hydrophobic stretch, 227PNVNC↓STRID, whose cleavage is probably mediated by signal peptidase (Fig. 6b). The second cleavage site was located at residues 435KRTKR↓WDSSY, as predicted above, which is the conserved motif recognized by furin-like proteases (Fig. 6b) [40]. Overall, the nascent DKNV ORF2a polypeptide could be directed to the endoplasmic reticulum and cleaved by signal peptidase and, in some circumstances, further cleaved by a furin-like protease in the Golgi complex to generate an N-terminally truncated spike glycoprotein (Fig. 6b). The two forms of putative spike glycoproteins, named msS1 and msS2, share a common C-terminus containing a TM region. The COILS [24] and Paircoil2 [25] programs predicted that the region followed by the putative furin cleavage site (that is, the msS2 domain) would have two coiled-coil structures (Fig. S2), which correspond to features that are functionally important for membrane fusion and entry of various viruses [e.g., 41, 42]. The calculated molecular masses of msS1 and msS2 are 77.9 kDa and 53.8 kDa, respectively, which are slightly smaller than the masses inferred from SDS gels (p80 and p58, respectively), probably due to lower electrophoretic mobility attributable to N-linked glycosylation (Fig. 6a, b), as is seen with other nidovirus spike glycoproteins [1]. In conclusion, the precursor DKNV ORF2a peptide is posttranslationally processed by a signal peptidase and/or a furin-like protease to express two types of outer spike glycoproteins with a common C-terminus (Fig. 6c). On the other hand, the N-terminal sequence of p30 could not be determined, probably because of a blocked N-terminal residue (Fig. 6a). The two minor polypeptides (p29 and p28) could not be subjected to N-terminal sequencing because of insufficient amounts of the polypeptides recovered, and it remains unclear whether the two polypeptides are incorporated into the DKNV virion. These issues should be addressed in future studies.
Phylogenetic study
To evaluate the taxonomic status of DKNV among the nidoviruses, we performed phylogenetic analysis based on RdRp sequences [26, 27]. Multiple sequence alignment of conserved motifs in nidovirus RdRp indicated that the DKNV RdRp sequence was most similar to that of members of the species Alphamesonivirus 1 (CavV and NDiV) in the genus Alphamesonivirus of the family Mesoniviridae [10], although there are some amino acid differences in conserved domains A and C between DNKV and members of Alphamesonivirus 1 (Fig. 7a). The resulting Bayesian phylogenetic tree showed that DKNV formed a clade with the viruses of Alphamesonivirus 1 (CavV and NDiV), clearly indicating that DKNV is a new member of the family Mesoniviridae (Fig. 7b). The topologies of NJ and ML trees were essentially the same as that of a Bayesian tree (Fig. S3). However, it should be noted that DKNV formed a separate branch from NDiV and CavV, which reflected the sequence differences in domains A and C.

Phylogenetic relationships between DKNV and other members of the order Nidovirales. (a) Alignment of the deduced amino acid sequences of the RdRp motif domains (F3, A, B, and C) [27, 28] from DKNV and 19 nidoviruses. Family or subfamily names of the viruses are indicated on the left side of the alignment. (b) The tree was constructed from the posterior distribution of trees generated by Bayesian MCMC coalescent analysis. Family or subfamily names are indicated in boldface type, and the viruses in the same families are circled. Posterior probabilities (on a scale from 0 to 1) are indicated above the branches. The scale bar represents the number of substitutions per site. Abbreviations of nidoviruses and sequence accession numbers are as follows: BCoV, bovine coronavirus ENT (NC_003045); BToV, bovine torovirus Breda1 (AY427798); EAV, equine arteritis virus Bucyrus (NC_002532); ECoV, equine coronavirus NC99 (NC_010327); EToV, equine torovirus Berne (X52374); GAV, gill-associated virus (NC_010306); HCoV-229E, human coronavirus 229E (NC_002645); HCoV-NL63, human coronavirus NL63 (NC_005831); IBV, infectious bronchitis virus Arkansas Vaccine (GQ504721); LDV, lactate dehydrogenase-elevating virus Plagemann (NC_001639); HHV, murine hepatitis virus A59 (AY700211); PEDV, porcine epidemic diarrhea virus CV777 (NC_003436); PRRSV, porcine respiratory and reproductive syndrome virus (NC_001961); SARS, SARS coronavirus Tor2 (NC_004718); SHFV, simian hemorrhagic fever virus LVR 42-0/M6941 (AF180391); WBV, white bream virus DF24/00 (NC_008516); YHV, yellow head virus Chachoengsao 1998 (EU487200)
Discussion
In this study, we characterized an insect nidovirus, DKNV, from mosquitoes of the species C. tritaeniorhynchus collected in the highlands of Vietnam. DKNV shared many characteristics with CavV [8] and NDiV [9], new members of the order Nidovirales that had also been discovered recently in mosquitoes. At present, these two nidoviruses are assigned to the same virus species, Alphamesonivirus 1, which is included in a new family, Mesoniviridae, based on the genomic features that are distinct from those of other known nidoviruses [10]. Our analyses confirm DKNV as a new member of the family Mesoniviridae.
Although these three nidoviruses share significant similarities in genome organization, DKNV showed subtle but significant differences from CavV and NDiV at the nucleotide and amino acid sequence levels. Amino acid sequence identities and similarities of six major ORFs (ORF1a, 1b, 2a, 2b, 3a, and 3b) among the three viruses suggested that CavV and NDiV are more closely related to each other than to DKNV (Table 1). In addition, the DKNV ORF1a protein was found to contain a 55-aa insertion (319K to 374G) and a 22-aa deletion (1720V to 1721S) compared with CavV and NDiV (Fig. S4). As a result, excluding the poly(A) tail, the genome of DKNV (20,256 bp) is slightly longer than those of CavV (20,108 bp) and NDiV (20,124 bp). The genomes of members of Alphamesonivirus 1 have been reported to contain six conserved active domains characteristic of nidoviruses – 3CL, RdRp, Zm-HEL, EXON, N-MT, and O-MT [10] – and such domains were also conserved in DKNV genome, sharing 75.8–89.5 % sequence identity with those of CavV and NDiV (Fig. S5). 5′-RACE analysis of the DKNV subgenomic RNAs identified two leader TRSs in the 5′-UTR of its genomic RNA (Fig. 4b). The DKNV TRS for subgenomic mRNA2 was almost identical to those of CavV and NDiV (Fig. S6). On the other hand, the DKNV TRS for subgenomic mRNA3 was identical to that of NDiV but not to CavV. Despite showing this partial discordance in TRSs, these three mesonidoviruses could use the same or a highly similar mechanism controlling the synthesis of 3′-coterminal subgenomic mRNAs. In conclusion, DKNV is deemed to be a new strain of the species Alphamesonivirus 1, and the current study provides further understanding of the viruses of the family Mesoniviridae.
This work has demonstrated that the mesonivirus ORF2a product undergoes posttranslational processing. The polypeptide encoded by the DKNV ORF2a is translated as a precursor polyprotein and is proteolytically processed into two forms of spike glycoprotein, msS1 and msS2, which share a common C-terminus containing two putative heptad repeat regions and a hydrophobic TM domain (Fig. 6 and Fig. S2). N-terminal sequencing of the virion structural proteins identified the two cleavage sites in the DKNV ORF2a polypeptide, 227PNVNC↓STRID and 435KRTKR↓WDSSY, which are probably cleaved by a signal peptidase and a furin-like protease, respectively. A similar pattern of spike glycoprotein precursor processing is found in other nidoviruses. The spike glycoprotein precursors of many coronaviruses have a signal peptide at the N-terminus and a TM domain at the C-terminus and, with few exceptions, become proteolytically cleaved by furin or a furin-like protease at a multibasic motif (often RRXRR↓) into two subunits [43, 44]. The spike glycoproteins of fish-infecting coronaviruses (genus Bafinivirus) possess a putative furin cleavage site at the motif R-X-K/R-R↓ [4]. The okavirus precursor glycoprotein contains a signal-peptide-like sequence close to the N-terminus, a proteolytic cleavage site, and a C-terminal TM domain. In this case, the second cleavage is probably mediated by signal peptidase, not a furin-like protease, just after the fifth TM domain close to the C-terminal side [45]. Interestingly, the ORF2a sequences of both NDiV and CavV also contain a putative furin-like cleavage motif (426 KRNKR for CavV and 425 KRSKR for NDiV) that is similar to the motif identified in DKNV ORF2a (435KRTKR↓), suggesting that the spike glycoprotein precursors of NDiV and CavV could also undergo post-translational processing. The two hypothetical spike glycoproteins of DKNV, msS1 (p80) and msS2 (p55), showed almost the same band intensity on SDS-PAGE (Fig. 6a), suggesting that the two forms of spike glycoproteins might be exposed on the virion surface in an approximately equal molar ratio.
The DKNV ORF2b was predicted to encode a nuclepcapsid protein because of its highly basicity and hydrophilicity (Fig. S1). In SARS-CoV, the nucleocapsid protein consists of two independent structural domains, an N-terminal RNA-binding domain and a C-terminal dimerization domain [46]. The ANCHOR server predicted that the putative nucleocapsid protein of DKNV possesses multiple disordered protein-binding regions in the N-terminal half and an ordered domain in the C-terminal half (Fig. S7). This predicted order/disorder pattern is very similar to those of the nucleocapsid proteins of CavV and NDiV. Interestingly, the amino acid sequence of the C-terminal half of the nucleocapsid protein is highly conserved among the three mesoniviruses (DKNV, CavV, and NDiV), in contrast to that of the N-terminal half (data not shown), implying that the C-terminal domain of mesonivirus nucleocapsid protein might be involved in RNA binding. In gill-associated virus (genus Okavirus, family Roniviridae), the RNA-binding domain of the nucleocapsid protein is located in its N-terminal region, which contains a proline/arginine-rich stretch [47]. Contrary to expectations, such a remarkable proline/basic residue-rich stretch was not found in any mesonivirus nucleocapsid proteins. Further studies are needed to elucidate the structural interaction between viral RNA and nucleocapsid protein in mesoniviruses.
DKNV could not replicate in the cultured vertebrate cells tested (Fig. 3), suggesting that DKNV is unlikely to be an infectious agent in vertebrate animals. In contrast, DKNV grew in all cultured mosquito cells tested. Interestingly, DKNV grew to high titers in two A. albopictus cell lines but grew poorly in a cell line derived from C. tritaeniorhynchus, which is the species from which DKNV was isolated. Suppressed and noncytopathic growth of DKNV in Culex mosquito cells suggests that in nature, DKNV establishes a latent infection in Culex mosquitoes as a natural reservoir host. Like DKNV, both CavV and NDiV were isolated from Culex mosquitoes, although CavV has also been isolated from Aedes mosquitoes. Recently, some flaviviruses associated with mosquitoes have been shown to be maintained in the natural population of mosquitoes by vertical transmission in both sexes [48, 49]. Is DKNV also maintained in the mosquito population by vertical transmission in nature? Zirkel et al. [8] suggested the presence of amplifying vertebrate hosts in the CavV infection cycle in nature because there was no evidence of CavV infection in male mosquitoes. To clarify how mesoniviruses are maintained in nature, more information about their host range, virulence, and tissue tropism should be gathered.
A new field of study, the new virus family Mesoniviridae, has just been opened up by the discoveries of CavV and NDiV. New information about the mesoniviruses themselves and related isolates, including DKNV, will make meaningful contributions not only to the ecological impact on mosquito populations, possibility for biological control of mosquitoes, and the evolutionary aspects of nidoviruses but also to the prevention of infection or antiviral drug development for nidovirus diseases of humans and animals.
References
de Groot RJ, Cowley JA, Enjuanes L, Faaberg KS, Perlman S, Rottier PJM, Snijder EJ, Ziebuhr J, Gorbalenya AE (2012) Order Nidovirales. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy; Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier, London, pp 785–795
Adams MJ, Carstens EB (2012) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Arch Virol 157:1411–1422
Schütze H, Ulferts R, Schelle B, Bayer S, Granzow H, Hoffmann B, Mettenleiter TC, Ziebuhr J (2006) Characterization of White bream virus reveals a novel genetic cluster of nidoviruses. J Virol 80:11598–11609
Batts WN, Goodwin AE, Winton JR (2012) Genetic analysis of a novel nidovirus from fathead minnows. J Gen Virol 93:1247–1252
Gorbalenya AE, Enjuanes L, Ziebuhr J, Snijder EJ (2006) Nidovirales: Evolving the largest RNA virus genome. Virus Res 117:17–37
Pasternak AO, Spaan WJ, Snijder EJ (2006) Nidovirus transcription: how to make sense? J Gen Virol 87:1403–1421
Junglen S, Kurth A, Kuehl H, Quan PL, Ellerbrok H, Pauli G, Nitsche A, Nunn C, Rich SM, Lipkin WI, Briese T, Leendertz FH (2009) Examining landscape factors influencing relative distribution mosquito genera and frequency of virus infection. Ecohealth 6:239–249
Zirkel F, Kurth A, Quan PL, Briese T, Ellerbrok H, Pauli G, Leendertz FH, Lipkin WI, Ziebuhr J, Drosten C, Junglen S (2011) An insect nidovirus emerging from a primary tropical rainforest. MBio 14:e00077-11
Nga PT, Parquet Mdel C, Lauber C, Parida M, Nabeshima T, Yu F, Thuy NT, Inoue S, Ito T, Okamoto K, Ichinose A, Snijder EJ, Morita K, Gorbalenya AE (2011) Discovery of the first insect nidovirus, a missing evolutionary link in the emergence of the largest RNA virus genomes. PLoS Pathog 7:e1002215
Lauber C, Ziebuhr J, Junglen S, Drosten C, Zirkel F, Nga PT, Morita K, Snijder EJ, Gorbalenya AE (2012) Mesoniviridae: a proposed new family in the order Nidovirales formed by a single species of mosquito-borne viruses. Arch Virol 157:1623–1628
Kuwata R, Nga PT, Yen NT, Hoshino K, Isawa H, Higa Y, Hoang NV, Trang BM, Loan DP, Phong TV, Hien NT, Sasaki T, Tsuda Y, Kobayashi M, Sawabe K, Takagi M (2006) Surveillance of Japanese Encephalitis Virus Infection in Mosquitoes in Vietnam from 2006 to 2008. Am J Trop Med Hyg 88:681–688
Hoshino K, Isawa H, Tsuda Y, Yano K, Sasaki T, Yuda M, Takasaki T, Kobayashi M, Sawabe K (2007) Genetic characterization of a new insect flavivirus isolated from Culex pipiens mosquito in Japan. Virology 359:405–414
Mitsuhashi J (1981) A new continuous cell line from larvae of the mosquito Aedes albopictus (Diptera Culicidae). Biochem Res 2:599–606
Kuwata R, Hoshino K, Isawa H, Tsuda Y, Tajima S, Sasaki T, Takasaki T, Kobayashi M, Sawabe K (2012) Establishment and characterization of a cell line from the mosquito Culex tritaeniorhynchus (Diptera: Culicidae). In Vitro Cell Dev Biol Anim 48:369–376
Igarashi A, Buei K, Ueba N, Yoshida M, Ito S, Nakamura H, Sasao F, Fukai K (1981) Isolation of viruses from female Culex tritaeniorhynchus in Aedes albopictus cell cultures. Am J Trip Med Hyg 30:449–460
Okuno Y, Igarashi A, Fukunaga T, Tadano M, Fukai K (1984) Electron microscopic observation of a newly isolated flavivirus-like virus from field-captured mosquitoes. J Gen Virol 65:803–807
Mizutani T, Endoh D, Okamoto M, Shirato K, Shimizu H, Arita M, Fukushi S, Saijo M, Sakai K et al (2007) Rapid genome sequencing of RNA viruses. Emerg Infect Dis 13:322–324
Sperschneider J, Datta A (2010) DotKnot: pseudoknot prediction using the probability dot plot under a refined energy model. Nucleic Acids Res 38:e103
Sperschneider J, Datta A, Wise MJ (2011) Heuristic RNA pseudoknot prediction including intramolecular kissing hairpins. RNA 17:27–38
Hofmann K, Stoffel W (1993) TMbase—A database of membrane spanning proteins segments. Biol Chem Hoppe-Seyler 374:166
Anders K, Bjorn L, Gunnar von H, Erik LLS (2001) Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol 305:567–580
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Duckert P, Brunak S, Blom N (2004) Prediction of proprotein convertase cleavage sites. Protein Eng Des Sel 17:107–112
Lupas A, Van Dyke M, Stock J (1991) Predicting coiled coils from protein sequences. Science 252:1162–1164
McDonnell AV, Jiang T, Keating AE, Berger B (2006) Paircoil2: Improved prediction of coiled coils from sequence. Bioinformatics 22:356–358
Dosztány Z, Mészáros B, Simon I (2009) ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics 15:2745–2746
Gorbalenya AE, Koonin EV (1993) Comparative analysis of amino acid sequences of the key enzymes of the replication and expression of positive-strand RNA viruses: validity of the approach and functional and evolutionary implications. SoV Sci Rev D Physicochem Biol 11:1–84
Beerens N, Selisko B, Ricagno S, Imbert I, van der Zanden L, Snijder EJ, Canard B (2007) De novo initiation of RNA synthesis by the arterivirus RNA-dependent RNA polymerase. J Virol 81:8384–8395
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Whelan S, Goldman N (2001) A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18:691–699
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282
Felsenstein J (2005) PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle
Felsenstein J (1985) Confidence-limits on phylogenies - an approach using the bootstrap. Evolution 39:783–791
Page RDM (2001) TreeView Version 1. 6. 6. University of Glasgow, Glasgow, UK. http://taxonomy.zoology.gla.ac.uk/rod/treeview.html
Ricagno S, Egloff MP, Ulferts R, Coutard B, Nurizzo D, Campanacci V, Cambillau C, Ziebuhr J, Canard B (2006) Crystal structure and mechanistic determinants of SARS coronavirus nonstructural protein 15 define an endoribonuclease family. Proc Natl Acad Sci USA 103:11892–11897
Bhardwaj K, Palaninathan S, Alcantara JM, Yi LL, Guarino L, Sacchettini JC, Kao CC (2008) Structural and functional analyses of the severe acute respiratory syndrome coronavirus endoribonuclease Nsp15. J Biol Chem 283:3655–3664
Nedialkova DD, Ulferts R, van den Born E, Lauber C, Gorbalenya AE, Ziebuhr J, Snijder EJ (2009) Biochemical characterization of arterivirus nonstructural protein 11 reveals the nidovirus-wide conservation of a replicative endoribonuclease. J Virol 83:5671–5682
Cowley JA, Cadogan LC, Spann KM, Sittidilokratna N, Walker PJ (2004) The gene encoding the nucleocapsid protein of Gill-associated nidovirus of Penaeus monodon prawns is located upstream of the glycoprotein gene. J Virol 78:8935–8941
Sittidilokratna N, Phetchampai N, Boonsaeng V, Walker PJ (2006) Structural and antigenic analysis of the yellow head virus nucleocapsid protein p20. Virus Res 116:21–29
Massova I, Kotra LP, Fridman R, Mobashery S (1998) Matrix metalloproteinases: structures, evolution, and diversification. FASEB J 12:1075–1095
Bosch BJ, van der Zee R, de Haan CA, Rottier PJ (2003) The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77:8801–8811
Chan WE, Chuang CK, Yeh SH, Chang MS, Chen SS (2006) Functional characterization of heptad repeat 1 and 2 mutants of the spike protein of severe acute respiratory syndrome coronavirus. J Virol 80:3225–3237
de Haan CA, Stadler K, Godeke GJ, Bosch BJ, Rottier PJ (2004) Cleavage inhibition of the murine coronavirus spike protein by a furin-like enzyme affects cell-cell but not virus-cell fusion. J Virol 78:6048–6054
de Haan CA, Haijema BJ, Schellen P, Wichgers Schreur P, te Lintelo E, Vennema H, Rottier PJ (2008) Cleavage of group 1 coronavirus spike proteins: how furin cleavage is traded off against heparan sulfate binding upon cell culture adaptation. J Virol 82:6078–6083
Jitrapakdee S, Unajak S, Sittidilokratna N, Hodgson RA, Cowley JA, Walker PJ, Panyim S, Boonsaeng V (2003) Identification and analysis of gp116 and gp64 structural glycoproteins of yellow head nidovirus of Penaeus monodon shrimp. J Gen Virol 84:863–873
Chang CK, Sue SC, Yu TH, Hsieh CM, Tsai CK, Chiang YC, Lee SJ, Hsiao HH, Wu WJ, Chang WL, Lin CH, Huang TH (2006) Modular organization of SARS coronavirus nucleocapsid protein. J Biomed Sci 13:59–72
Soowannayan C, Cowley JA, Michalski WP, Walker PJ (2011) RNA-binding domain in the nucleocapsid protein of gill-associated nidovirus of penaeid shrimp. PLoS One 6:e22156
Bolling BG, Eisen L, Moore CG, Blair CD (2011) Insect-specific flaviviruses from Culex mosquitoes in Colorado, with evidence of vertical transmission. Am J Trop Med Hyg 85:169–177
Saiyasombat R, Bolling BG, Brault AC, Bartholomay LC, Blitvich BJ (2011) Evidence of efficient transovarial transmission of Culex flavivirus by Culex pipiens (Diptera: Culicidae). J Med Entomol 48:1031–1038
Acknowledgements
The authors would like to thank Dr. Masahiro Takagi of Nagasaki University for helpful discussion, and Dr. Yukiko Higa of Nagasaki University and the staff members of Department of Medical Entomology and Zoology, National Institute of Hygiene and Epidemiology, for their arrangements for field work. We also thank Drs. Kikuo Iwabuchi of Tokyo University of Agriculture and Technology and Tohru Yanase of Kyushu Research Station, National Institute of Animal Health, NARO, for providing NIAS-AeAl-2 and HmLu-1 cells, respectively. This work was supported in part by a grant-in-aid from the Japanese Ministry of Health, Labor and Welfare (H24-Shinko-Ippan-007) and JSPS KAKENHI grant numbers 21406012, 22590387 and 24790134.
Author information
Authors and Affiliations
Corresponding author
Additional information
R. Kuwata and T. Satho contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Kuwata, R., Satho, T., Isawa, H. et al. Characterization of Dak Nong virus, an insect nidovirus isolated from Culex mosquitoes in Vietnam. Arch Virol 158, 2273–2284 (2013). https://doi.org/10.1007/s00705-013-1741-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-013-1741-4