
Abstract
Recent advances have shown that atypical hemolytic uremic syndrome (aHUS) is a disease of complement dysregulation. Almost 50% of cases are associated with mutations in the three complement regulatory genes, factor H (HF1), membrane co-factor protein (MCP) and factor I (IF). The corresponding gene products act in concert and affect the same enzyme, alternative pathway convertase C3bBb, which initiates the alternative pathway and amplification of the complement system. Factor H (FH) deficiency-associated aHUS usually occurs in infants to middle-aged adults and only rarely in neonates. Moreover, the vast majority of patients are heterozygous for the HF1 gene mutations. We report on a case of neonatal-onset aHUS associated with complete FH deficiency due to novel compound heterozygous mutations in the HF1 gene. A 22-day-old baby girl developed acute renal failure and a remarkably low serum complement C3 level, which was rapidly followed by the development of micro-angiopathic hemolytic anemia. Western blot analysis revealed nearly zero plasma FH levels, and an HF1 gene study showed compound heterozygous mutations, C1077W/Q1139X. Renal pathology findings were compatible with glomerular involvement in HUS. The baby recovered completely after the repetitive infusion of fresh frozen plasma. During follow-up (until she was 20 months old) after the initial plasma therapy, the disease recurred three times; twice after the tapering off of plasma therapy, and once during a weekly plasma infusion. All recurrence episodes were preceded by an upper respiratory tract infection, and were successfully managed by restarting or increasing the frequency of plasma therapy.
Similar content being viewed by others

Introduction
Hemolytic uremic syndrome (HUS) is a clinical syndrome characterized by micro-angiopathic hemolytic anemia, thrombocytopenia, and acute renal failure. In most cases, HUS follows a prodrome of diarrhea (D+ HUS) typically caused by Shiga toxin-producing Escherichia coli [1]. Atypical HUS (aHUS) is a rare, non-Shiga toxin-associated HUS, which affects a heterogeneous group of patients and shows tendencies to relapse, to occur in families, and to have a poor outcome [1–3]. aHUS is a disease of complement dysregulation, and mutations in genes coding for complement regulatory proteins, such as complement factor H (FH) [4–11], membrane co-factor protein (MCP/CD46) [11–14] and factor I [11, 15, 16] are detected in almost half of affected patients. Each of these three gene proteins control the same enzyme, namely, alternative C3 convertase C3bBb [17].
The FH gene (HF1) was the first gene shown to be linked to aHUS [4–11], and it has also been associated with other renal diseases, such as, type II membranoproliferative glomerulonephritis (MPGN II) [10, 18, 19] and type III collagen glomerulopathy [20–23]. To date, more than 65 mutations in the HF1 gene have been reported in aHUS patients [24], and most of these have been heterozygous. Moreover, FH deficiency-associated aHUS can develop at any age, but it is rare in neonates [5, 10, 25].
In this paper we describe a case of neonatal-onset aHUS with total deficiency of plasma FH due to compound heterozygous mutations in the HF1 gene.
Case report
The patient was a Korean female infant born at gestation week 40 with a birth weight of 3,480 g to a mother with a previous history of spontaneous abortion of unknown etiology. She was the first child of non-consanguineous parents, and her antenatal and perinatal histories were unremarkable. At age 22 days, a few days after a BCG vaccination, she developed generalized edema accompanied by vomiting, oliguria, gross hematuria and proteinuria (3+), and during the following 3 days her weight increased from 4,000 g to 5,070 g. On admission, at age 25 days, her pulse rate was 160/min, respiratory rate 70/min, and blood pressure 140/90 mmHg. Initial laboratory tests revealed blood urea nitrogen 45 mg/dl, serum creatinine 1.9 mg/dl, serum albumin 2.6 g/dl, hemoglobin 9.3 g/dl, and platelets 188,000/ul. Urinalysis showed hematuria and albuminuria (3+), and her random urine protein (milligram/deciliter) to creatinine (milligram/deciliter) ratio (urine prot/cr) was 23.3. On the third hospital day, follow-up laboratory tests revealed definite evidence of micro-angiopathic hemolytic anemia, i.e., hemoglobin 8.0 g/dl, reticulocyte count 4.7%, platelets 89,000/ul, many schistocytes in a peripheral smear, and negative results of direct and indirect Coombs’ tests. Serum C3 and C4 levels were 8 mg/dl and 9 mg/dl, respectively, and findings of her fluorescence anti-nuclear antibody test were negative. Renal Doppler ultrasonography showed increased parenchymal echogenicity of both kidneys without evidence of renal vein thrombosis. Because of uncontrollable edema and hypertension, peritoneal dialysis started on the second hospital day, at age 26 days, and 20% albumin and packed red cells were infused intermittently. Dialysis was discontinued after 3 weeks, when platelets (>200,000/ul), renal function (serum creatinine 0.4 mg/dl), and urine output recovered spontaneously. Nevertheless, at this time, hypertension, low C3, a mild degree of hemolytic anemia, and urinary abnormalities (urine protein/creatinine 12.1 and microscopic hematuria) persisted.
When she was 2 months of age, oliguria and azotemia (serum creatinine 1.2 mg/dl) recurred, and a renal biopsy, plasma FH level (by western blot) and an HF1 gene study were performed. After the confirmation of FH deficiency, fresh frozen plasma (FFP) infusion (15 ml/kg body weight, three times a week) was started, and, 7 weeks after the first FFP infusion, she was discharged with a completely normalized blood pressure, peripheral blood smear, and renal function, except intermittent mild proteinuria.
After discharge, FFP infusions were tapered off slowly, i.e., weekly for the first 2 months, on alternate weeks for another 1 month, and then stopped for 2 months, during which all laboratory test results remained normal, except for a persistent low C3 level. However, when she was aged 9 months, hypertension, gross hematuria, and oliguria re-emerged a few days after an upper respiratory tract infection. Laboratory test results at the time included hemoglobin 5.9 g/dl, platelets 38,000/ul, serum creatinine 2.0 mg/dl, blood urea nitrogen 105 mg/dl, and massive proteinuria (urine protein/creatinine 38.6). Thus, FFP infusion therapy was restarted, and she recovered within 3 weeks, but the proteinuria persisted (urine protein/creatinine 2.9). FFP infusions were then slowly tapered off again, and she remained well for 4 months without FFP thereafter. Two doses of diphtheria–pertussis–tetanus (DPT) vaccine (regular immunization) were given without problem when she was 15 months and 17 months, respectively. When she was 18 months and 19 months old, third and fourth relapses developed, with concomitant upper respiratory tract infections. The fourth relapse occurred during weekly FFP therapy. Both episodes were successfully managed by restarting or increasing frequency of plasma therapy. Her serial serum C3 levels were 16 mg/dl to 59 mg/dl during FFP therapy and 5 mg/dl to 10 mg/dl without FFP. At the last follow-up, she was 20 months old with normal growth and development, and her renal function remained normal except for mild proteinuria (urine protein/creatinine 0.6–1.9)
Materials and methods
Plasma FH levels were measured by western blotting with an ECL kit (Amersham Life Science, Buckinghamshire, UK) as previously described [26].
For genetic analysis of the HF1 gene, genomic DNA was isolated from the peripheral blood nucleated cells of the patient and her parents. All coding exons and flaking introns of the HF1 gene were amplified from genomic DNA by polymerase chain reaction (PCR) and were directly sequenced.
In addition to routine light, immunofluorescence and electron microscopy examinations, a portion of a renal biopsy specimen was stained for type III collagen using a polyclonal rabbit anti-human type III collagen antibody (Biodesign International, Saco, Maine, USA) as a primary antibody and fluorescein-labeled anti-rabbit IgG antibody as a secondary antibody.
Result
The baseline plasma FH level of this patient was 3% of normal (Fig. 1, upper panel). Serial plasma FH levels were measured when the patient was on FFP therapy of 2-week interval after the initial episode. After FFP infusion (15 ml/kg body weight), plasma FH increased to 25% of normal on day 1 and dropped to 13% and 6% of normal on days 4 and 7, respectively (Fig. 1, lower panel). Plasma FH levels of both parents were approximately half of normal, but serum C3 levels were normal.

Plasma FH levels of family members measured by western blotting (upper panel). Lane 1 the patient (3% of normal); lane 2 her mother (41% of normal); lane 3 her father (48% of normal); lanes 4 and 5 healthy controls. Serial measurement of plasma FH levels in the patient after fresh frozen plasma infusion (15 ml/kg body weight) (lower panel). Lane 1 a healthy control; lane 2 blank; lane 3 just before infusion (3% of normal); lane 4 just after infusion (22% of normal); lane 5 1 day after infusion (25% of normal); lane 6 4 days after infusion (13% of normal); lane 7 7 days after infusion (6% of normal)
Two novel heterozygous mutations were detected in the HF1 gene of the patient; a T>G point mutation, replacing the 1077 cysteine residue with tryptophan in exon 21 [in the 18th short consensus repeats (SCRs) domain), and a C>T point mutation, replacing 1139 glutamine with a premature stop codon in exon 22 (in the 19th SCR domain). The patient had inherited the former mutation from her mother and the latter from her father (Fig. 2).

Partial HF1 gene sequencing data. A C>T heterozygous nonsense point mutation of maternal origin and a T>G heterozygous missense point mutation of paternal origin were detected in exons 22 and 21, respectively
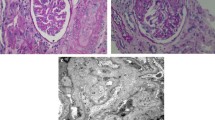
A light microscopy examination of a renal biopsy revealed marked mesangial and endothelial cell proliferation and a mesangial matrix with a fibrillar appearance. Double-contoured glomerular capillary loops were observed focally (Fig. 3). Nine of 132 glomeruli exhibited cellular crescent formation, and some glomeruli and tubules had an immature appearance. Immunofluorescence studies showed peripheral and mesangial staining for C3 and fibrinogen (Fig. 4), and an indirect immunofluorescence study demonstrated no staining for type III collagen (data not shown). Electron microscopy demonstrated focal subendothelial widening and mesangial interposition without an electron-dense deposit. Some fibrils existed focally in the mesangium without showing periodicity (Fig. 5).

Light microscopy examination of a renal biopsy taken when the patient was 2 months old. Marked mesangial and endothelial hypercellularity was noted, and the mesangial matrix had a fibrillar appearance. Double-contoured glomerular capillary loops were observed focally. Periodic acid–Schiff staining, ×400)

Immunofluorescence studies of a renal biopsy taken when the patient was 2 months old showed peripheral and mesangial staining for C3 and fibrinogen

Electron microscopy showed focal subendothelial widening (asterisks) without an electron-dense deposit. Some focally distributed fibrils (arrows) were observed in the mesangium but showed no periodicity (×2,000)
Discussion
FH is a 150-kDa serum glycoprotein that is predominantly synthesized by the liver, and it is composed of 20 tandem-arrayed homologous units of approximately 60 amino acids, which are called a SCR or complement control protein. FH acts as an essential control protein of the alternative complement pathway by: (1) functioning as a co-factor for the factor I-mediated proteolytic inactivation of C3b, (2) competing with factor B for C3b binding, and (3) accelerating the decay of C3 convertase into its components. An FH molecule possesses three C3-binding sites and three heparin-binding sites [2, 17, 24, 27].
Inherited FH deficiency leads to the chronic uncontrolled activation of the alternative complement pathway, and it can cause several types of renal disease, such as aHUS [4–11, 25, 26], MPGN II [10, 18, 19] and collagen type III glomerulopathy [20–23]. FH deficiencies have been reported in both homozygous and heterozygous genotypic forms or as complete and partial forms based on antigenic or functional plasma levels. The majority of aHUS patients harbor heterozygous HF1 mutations, which cause partial deficiency. However, some show complete FH deficiency and homozygous [5, 10, 11, 18, 28] or compound heterozygous mutations [6, 7, 9, 26, 29, 30]. In contrast, all patients with MPGN II show complete or homozygous deficiency [10, 18]. It remains unclear whether the observed renal histologic variabilities in patients with FH deficiency are associated with different pathogenetic mechanisms, and why some individuals with heterozygous FH deficiency remain free of apparent disease. In our patient, plasma FH level findings revealed complete deficiency, and gene analysis showed compound heterozygous mutations in the HF1 gene. A final diagnosis of aHUS was made in our patient based on the lack of dense deposits within the glomerular basement membranes (the morphological hallmark of MPGN II) and type III collagen deposition. However, the renal pathology of our patient was rather unusual for aHUS, because glomerular capillaries were predominantly affected. In general, this glomerular pattern is typical of D+ HUS and differs from the pre-glomerular pattern of aHUS, which predominantly affects renal arterioles and interlobular arteries [2, 3].
Two novel heterozygous mutations in the HF1 gene were detected in our patient: C1077W in SCR 18 and Q1139X in SCR 19. Each SCR in the FH molecule contains four cysteine residues, which play an essential role during protein folding by forming disulfide bridges [23]. Many of the reported missense point mutations in the FH gene involve the substitution of one of these conserved cysteine residues [17, 23], e.g., C1077W in our patient. Such mutant proteins are known to be retained intracellularly and not secreted to plasma [27]. On the other hand, Q1139X is a nonsense mutation that produces a stop codon, and it is suggested that the truncated protein produced is also retained intracellularly, because it, too, was not detected in plasma. HF1 mutations associated with aHUS in the literature, such as Q1139X in our patient, cluster in the C-terminal domain of the FH molecule (SCRs 19 and 20) [2, 17]. The C-terminal domain is the central site for surface recognition and has binding sites for polyanions and C3b [2, 17].
Mutations in genes coding for FH, MCP or factor I have been found in aHUS patients [4–16], but incomplete penetrance of the disease in individuals carrying these mutations is relatively frequent and no genetic defect has yet been found in about a half of patients [2]. Recently, a large aHUS pedigree was reported [14] in which independent segregation of three different aHUS risk factors was detected [a missense mutation in the MCP gene (MCP), a frame-shifting dinucleotide insertion in the factor I gene (IF), and a specific MCP single-nucleotide polymorphism haplotype block], and only individuals carrying the three aHUS risk factors were affected by aHUS. These data help to explain the incomplete penetrance of the disease by illustrating that concurrence of multiple hits in complement regulatory proteins may be necessary for development of aHUS. In our patient, although compound heterozygous mutations in HF1 were detected, other genes, including MCP and IF, were not studied. Thus, possible involvement of other genetic risk factor(s) cannot be excluded completely.
FH deficiency-associated aHUS may develop at any age, but it is rare in neonates [5, 10, 25]. In most patients harboring heterozygous mutations, aHUS develops during late infancy or adulthood and is often triggered by factors such as infection or pregnancy [5]. Thus, it is suggested that, in those with FH haplo-insufficiency, other environmental or acquired factors are required for disease manifestation. In contrast, homozygous or compound heterozygous mutations result in severe reductions in plasma FH, and they are accompanied by early onset disease, even in the absence of a triggering factor [5]. In a recent study, 70% of cases of FH deficiency-associated aHUS were found to have been preceded by an infection and 4% by pregnancy or drug administration [11]. In our patient, aHUS onset was preceded by a BCG vaccination, and three of four relapses were precipitated by upper respiratory tract infections. This effect of a BCG vaccination is novel, although one case of aHUS reportedly followed a measles–mumps–rubella vaccination [31], and another patient with a homozygous FH deficiency relapsed after a pneumococcal vaccination [28].
Although no prospective randomized controlled trials have confirmed their efficacies, plasma exchange and plasma infusion are recognized first line therapies for aHUS, and the adoption of these modalities has been credited with a reduction in overall mortality of from 50% to 25% [3]. It has also been suggested that plasma exchange may be more effective than plasma infusion alone, because of the larger quantity involved and the associated removal of possible toxic substances. Moreover, response to plasma therapy is a function of genetic background. In a recent large-scale study [11], 93% of episodes (in patients with HF1 mutations) were treated with plasma and 67% achieved remission. Similar results were observed in patients with IF mutations. In contrast, plasma therapy was used to treat 66% of acute episodes in patients with MCP mutations, and, although remission was achieved in 91% of plasma-treated episodes, it was also obtained in 100% of non-treated episodes. These findings suggest that plasma does not have a significant impact on the outcome of aHUS associated with MCP mutations. This is probably due to the fact that MCP (unlike FH and factor I, which are circulating proteins) is a membrane-bound protein, and, thus, plasma therapy would not be expected to correct an MCP defect [11].
It is difficult to decide how long and often plasma therapy should be administered after recovery from an acute attack. In a case report of homozygous FH deficiency-associated aHUS [28], the plasma half-life of FH after FFP infusion (20 ml/kg body weight) was determined to be approximately 6 days, which provides a scientific basis for the commonly recommended 2-week interval for maintenance plasma therapy. However, individual responses to plasma therapy vary, even between identical twins [32], and patients occasionally experience substantial remission periods after an initial attack without FFP infusion. In the present study we measured serial plasma FH levels after FFP infusion and found that peak FH level after infusion reduced by approximately 50% after 4 days. However, we were unable to calculate accurately the half-life of infused FH, because western blotting, rather than enzyme-linked immunosorbent assay (ELISA), was used, and too few measurements were taken. In addition, we were unable to continue FFP infusion at 2-week-intervals, due to the withdrawal of parental consent. However, one relapse occurred during weekly FFP therapy, which suggests that 2-week-intervals may have been insufficient.
The overall long-term prognosis of aHUS is problematic. The majority of patients experience multiple relapses and eventually progress to end-stage renal disease. Approximately 50% of patients who undergo renal transplantation experience aHUS recurrence [33–36], and more than 90% of these patients progress to graft failure [11]. Recurrence rates in patients with HF1 and IF mutations are ca. 80% [37] and 100% [11, 15, 16], respectively. In contrast, in patients with MCP mutations, recurrence occurs in fewer than 10% [11, 12, 37]. Because FH is a protein of mainly hepatic origin, liver transplantation with/without kidney transplantation has been attempted recently with curative intent. However, the results of three initial reports [25, 26, 38] were disappointing, as two of these patients died. Nevertheless, a recent report [30] described a successful treatment involving preoperative plasma exchange. It was suggested that preoperative plasma therapy provides the recipient with functioning FH, which prevents local complement activation in grafts immediately after transplantation [30].
In summary, we describe a rare case of neonatal onset aHUS with a glomerular renal pathology pattern and complete FH deficiency due to novel compound heterozygous mutations in the HF1 gene.
References
Ruggenenti P, Noris M, Remuzzi G (2001) Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int 60:831–846
Kavanagh D, Goodship THJ, Richards A (2006) Atypical haemolytic uraemic syndrome. Br Med Bull 77–78:5–22
Noris M, Remuzzi G (2005) Non-Shiga toxin-associated hemolytic uremic syndrome. In: Zipfel PF (ed) Complement and kidney disease. Birkhäuser, pp 65–83
Warwicker P, Goodship THJ, Donne RL, Pirson Y, Nicholls A, Ward RM, Turnpenny P, Goodship JA (1998) Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int 53:836–844
Caprioli J, Bettinaglio P, Zipfel PF, Amadei B, Daina E, Gamba S, Skerka C, Marziliano N, Remuzzi G, Noris M (2001) The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Am Soc Nephrol 12:297–307
Perez-Caballero D, Gonzalez-Rubio C, Gallardo ME, Vera M, Lopez-Trascasa M, Rodriguez de Cordoba S, Sanchez-Corral P (2001) Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet 68:478–484
Richards A, Buddles MR, Donne RL, Kaplan BS, Kirk E, Venning MC, Tielemans CL, Goodship JA, Goodship TH (2001) Factor H mutations in hemolytic uremic syndrome cluster in exons 18–20, a domain important for host cell recognition. Am J Hum Genet 68:485–490
Caprioli J, Castelletti F, Bucchioni S, Bettinaglio P, Bresin E, Pianetti G, Gamba S, Brioschi S, Daina E, Remuzzi G, Noris M, International Registry of Recurrent and Familial HUS/TTP (2003) Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet 12:3385–3395
Neumann HP, Salzmann M, Bohnert-Iwan B, Mannuelian T, Skerka C, Lenk D, Bender BU, Cybulla M, Riegler P, Konigsrainer A, Neyer U, Bock A, Widmer U, Male DA, Franke G, Zipfel PF (2003) Haemolytic uraemic syndrome and mutations of the factor H gene: a registry-based study of German speaking countries. J Med Genet 40:676–681
Dragon-Durey MA, Fremeaux-Bacchi V, Loirat C, Blouin J, Niaudet P, Deschenes G, Coppo P, Herman Fridman W, Weiss L (2004) Heterozygous and homozygous factor H deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: report and genetic analysis of 16 cases. J Am Soc Nephrol 15:787–795
Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, Mele C, Bresin E, Cassis L, Gamba S, Porrati F, Bucchioni S, Monteferrante G, Fang CJ, Liszewski MK, Kavanagh D, Atkinson JP, Remuzzi G, International Registry of Recurrent and Familial HUS/TTP (2006) Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 108:1267–1279
Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, Decorte R, Muslumanoglu MH, Kavukcu S, Filler G, Pirson Y, Wen LS, Atkinson JP, Goodship TH (2003) Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci USA 100:12966–12971
Noris M, Brioschi S, Caprioli J, Todeschini M, Bresin E, Porrati F, Gamba S, Remuzzi G, International Registry of Recurrent and Familial HUS/TTP (2003) Familial haemolytic uraemic syndrome and an MCP mutation. Lancet 362:1542–1547
Esparza-Gordillo J, Jorge EG, Garrido CA, Carreras L, Lopez-Trascasa M, Sanchez-Corral P, de Cordoba SR (2006) Insights into hemolytic uremic syndrome: segregation of three independent predisposition factors in a large, multiple affected pedigree. Mol Immunol 43:1769–1775
Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, Vigneau C, Kuypers D, Boudailliez B, Loirat C, Rondeau E, Fridman WH (2004) Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet 41:e84–e88
Kavanagh D, Kemp EJ, Mayland E, Winney RJ, Duffield JS, Warwick G, Richards A, Ward R, Goodship JA, Goodship TH (2005) Mutations in complement factor I predispose to development of atypical hemolytic uremic syndrome. J Am Soc Nephrol 16:2150–2155
Zipfel PF, Skerka C (2006) Complement dysfunction in hemolytic uremic syndrome. Curr Opin Rheumatol 18:548–555
Licht C, Heinen S, Józsi M, Löschmann I, Saunders RE, Perkins SJ, Waldherr R, Skerka C, Kirschfink M, Hoppe B, Zipfel PF (2006) Deletion of Lys224 in regulatory domain 4 of factor H reveals a novel pathomechanism for dense deposit disease (MPGN II). Kidney Int 70:42–50
Licht C, Schlötzer-Schrehardt U, Kirschfink M, Zipfel PF, Hoppe B (2007) MPGN II—genetically determined by defective complement regulation? Pediatr Nephrol 22:2–9
Imbasciati E, Gherardi G, Morozumi K, Gudat F, Epper R, Basler V, Mihatsch MJ (1991) Collagen type III glomerulopathy: a new idiopathic glomerular disease. Am J Nephrol 11:422–429
Vogt BA, Wyatt RJ, Burke BA, Simonton SC, Kashtan CE (1995) Inherited factor H deficiency and collagen type III glomerulopathy. Pediatr Nephrol 9:11–15
Bircan Z, Toprak D, Kilicaslan I, Solakoglu S, Uysal V, Ponard D, Turker G (2004) Factor H deficiency and fibrillary glomerulopathy. Nephrol Dial Transplant 19:727–730
Ault B, Schmidt B, Fowler N, Kashtan C, Ahmed A, Vogt B, Colten HR (1997) Human factor H deficiency: mutations in framework cysteine residues and block in H protein secretion and intracellular catabolism. J Biol Chem 272:25168–25175
Skerka C, Józsi M (2006) Role of complement and factor H in haemolytic uraemic syndrome. In: Zipfel PF (ed) Complement and kidney diseases. Birkhäuser, pp 85–109
Remuzzi G, Ruggenenti P, Codazzi D (2002) Combined kidney and liver transplantation for familial haemolytic uraemic syndrome. Lancet 359:1671–1672
Cheong HI, Lee BS, Kang HG, Hahn H, Suh K-S, Ha IS, Cho Y (2004) Attempted treatment of factor H deficiency by liver transplantation. Pediatr Nephrol 19:454–458
Zipfel PF, Misselwitz J, Licht C, Skerka C (2006) The role of defective complement control in hemolytic uremic syndrome. Semin Thromb Hemost 32:146–154
Licht C, Weyersberg A, Heinen S, Stapenhorst L, Devenge J, Beck B, Waldherr R, Kirschfink M, Zipfel PF, Hoppe B (2005) Successful plasma therapy for atypical hemolytic uremic syndrome caused by factor H deficiency owing to a novel mutation in the complement cofactor protein domain 15. Am J Kidney Dis 45:415–421
Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, Carreras Berges L, Lopez-Trascasa M, Sanchez-Corral P, Rodriguez de Cordoba S (2005) Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet 14:703–712
Saland JM, Emre SH, Shneider BL, Benchimol C, Ames S, Bromberg JS, Remuzzi G, Strain L, Goodship THJ (2006) Favorable long-term outcome after liver–kidney transplant for recurrent hemolytic uremic syndrome associated with a factor H mutation. Am J Transplant 6:1948–1952
Karim Y, Masood A (2002) Haemolytic uraemic syndrome following mumps, measles, and rubella vaccination. Nephrol Dial Transplant 17:941–942
Davin J-C, Olie KH, Verlaak R, Horuz F, Florquin S, Weening JJ, Groothoff JW, Strain L, Goodship THJ (2006) Complement factor H-associated atypical hemolytic uremic syndrome in monozygotic twins: concordant presentation, discordant response to treatment. Am J Kidney Dis 47:227–230, 2006
Artz MA, Steenbergen EJ, Hoitsma AJ, Monnens LA, Wetzels JF (2003) Renal transplantation in patients with hemolytic uremic syndrome: high rate of recurrence and increased incidence of acute rejections. Transplantation 76:821–826
Miller RB, Burke BA, Schmidt WJ, Gillingham KJ, Matas AJ, Mauer M, Kashtan CE (1997) Recurrence of haemolytic–uraemic syndrome in renal transplants: a single-centre report. Nephrol Dial Transplant 12:1425–1430
Loirat C, Niaudet P (2003) The risk of recurrence of hemolytic uremic syndrome after renal transplantation in children. Pediatr Nephrol 18:1095–1101
Zimmerhackl LB, Scheiring J, Prüfer F, Taylor CM, Loirat C (2007) Renal transplantation in HUS patients with disorders of complement regulation. Pediatr Nephrol 22:10–16
Kavanagh D, Goodship T (2006) Membrane cofactor protein, factor I mutations and transplantation. Semin Thromb Hemost 32:155–159
Remuzzi G, Ruggenenti P, Colledan M, Gridelli B, Bertani A, Bettinaglio P, Bucchioni S, Sonzogni A, Bonanomi E, Sonzogni V, Platt JL, Perico N, Noris M (2005) Hemolytic uremic syndrome: a fatal outcome after kidney and liver transplantation performed to correct factor H gene mutation. Am J Transplant 5:1146–1150
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cho, H.Y., Lee, B.S., Moon, K.C. et al. Complete factor H deficiency-associated atypical hemolytic uremic syndrome in a neonate. Pediatr Nephrol 22, 874–880 (2007). https://doi.org/10.1007/s00467-007-0438-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-007-0438-x