
Abstract
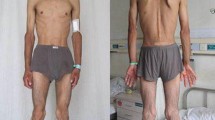
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a rare autosomal recessive disorder in which a nuclear mutation of the thymidine phosphorylase (TP) gene causes mitochondrial genomic dysfunction. Patients suffer from gastrointestinal dysmotility, cachexia, ptosis, external ophthalmoparesis, myopathy and polyneuropathy. Magnetic resonance imaging (MRI) shows leukoencephalopathy. We describe clinical, genetic and neuroradiological features of three brothers affected with MNGIE. Clinical examination, laboratory analyses, MRI and magnetic resonance spectroscopy (MRS) of the brain, and genetic analysis have been performed in all six members of the family with the three patients with MNGIE. Two of them are monozygous twins. They all suffered from gastrointestinal dysmotility, cachexia, ophthalmoplegia, muscular atrophies, and polyneuropathy. Urinary thymidine was elevated in the patients related to the severity of clinical disease, and urinary thymidine (normally not detectable) was also found in a heterozygous carrier. Brain MRI showed leukoencephalopathy in all patients; however, their cognitive functioning was normal. Brain MRS demonstrated reduced N-acetylaspartate and choline in severely affected areas. MRI of heterozygous carriers was normal. A new mutation (T92N) in the TP gene was identified. Urinary thymidine is for the first time reported to be detectable in a heterozygous carrier. MRS findings indicate loss of neurons, axons, and glial cells in patients with MNGIE, but not in heterozygous carriers.



Similar content being viewed by others


References
Okamura K, Santa T, Nagae K, et al. (1976) Congenital okuloskeletal myopathy with abnormal muscle and liver mitochondria. J Neurol Sci 27:79–91
Ionasescu V (1983) Oculogastrointestinal muscular Dystrophy. Am J Med Genet 15:103–112
Cervera R, Bruix J, Bayes A, et al. (1988) Chronic intestinal pseudoobstruction and ophthalmoplegia in a patient with mitochondrial myopathy. Gut 29:544–547
Threlkeld AB, Miller NR, Golnik KC, et al. (1992) Ophthalmic involvement in myo-neuro-gastrointestinal encephalopathy syndrome. Am J Ophthal 114:322–328
Simon LT, Horoupian DS, Dorfman LJ, et al. (1990) Polyneuropathy, ophthalmoplegia, leukoencephalopathy, and intestinal pseudo-obstruction: POLIP syndrome. Ann Neurol 28:349–360
Faber J, Fich A, Steinberg A, et al. (1987) Familial intestinal pseudoobstruction dominated by progressive nbeurologic disease at a young age. Gastroenterology 92:786–790
Bardosi A, Creutzfeldt W, DiMauro S, et al. (1987) Myo-, neuro-, gastrointestinal encephalopathy (MNGIE syndrome) due to a partial deficiency of cytochrome-c-oxidase. Acta Neuropathol (Berlin) 74:248–258
Teitelbaum JE, Berde CB, Nurko S, et al. (2002) Diagnosis and management of MNGIE syndrome in children: Case report and review of the literature. J Pediatr Gastroenterol Nutr 35:377–383
Uncini A, Servidei S, Silvestri G, et al. (1994) Ophthalmoplegia, demyelinating neuropathy, leukoencephalopathy,myopathy, and gastrointestinal dysfunction with multiple deletions of mitochondrial DNA: A mitochondrial multisystem disorder in search of a name. Muscle Nerve 17:667–674
Blake D, Lombes A, Minetti C, et al. (1990) MNGIE syndrome: Report of 2 new patients [abstract]. Neurology 40(suppl 1):294(643S)
Debouverie M, Wagner M, Ducrocq X, et al. (1997) Le MNGIE syndrome: deux cas dans une même fratrie. Rev Neurol (Paris) 153:547–553
Steiner I, Steinberg A, Argov Z, et al. (1987) Familial progressive neuronal disease and chronic idiopathic intestinal pseudo-obstruction. Neurology 37:1046–1050
Papadimitriou A, Comi GP, Hadjigeorgiou GM, et al. (1998) Partial depletion and multiple deletions of muscle mtDNA in familial MNGIE syndrome. Neurology 51:1086–1092
Hirano M, Silvestri G, Blake DM, et al. (1994) Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): Clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology 44:721–727
Sabatelli M, Servidei S, Ricci E, et al. (1992) Myo-neuro-gastro-intestinal disease and encephalopathy (MNGIE syndrome): a patient with multiple deletions of mitochondrial DNA [abstract]. Neurology 42(suppl 3):418
Carrozzo R, Hirano M, Fromenty B, et al. (1998) Multiple mtDNA deletions features in autosomal dominant and recessive diseases suggest distinct pathogeneses. Neurology 50:99–106
Cave DR (1990) Case records of the Massachusetts General Hospital, Case 12–1990. New Engl J Med 322:829–841
Mueller LA, Camilleri M, Emslie-Smith AM (1999) Mitochondrial neurogastrointestinal encephalomyopathy: Manometric and diagnostic features. Gastroenterology 116:959–963
Lowsky R, Davidson G, Wolman S, et al. (1993) Familial visceral myopathy associated with a mitochondrial myopathy. Gut 34:279–283
Nishino I, Spinazzola A, Papadimitriou A, et al. (2000) Mitochondrial Neurograstrointestinal Encephalomyopathy: An Autosomal Recessive Disorder due to Thymidine Phosphorylase Mutations. Ann Neurol 47:792–800
Labague P, Durant R, Castelnuovo G, et al. (2002) MNGIE: Diarrhea and leukoencephalopathy. Neurology 58:1862
Gamez J, Ferreiro C, Accarino ML, et al. (2002) Phenotypic variability in a Spanish family with MNGIE. Neurology 59:455–457
Hamano H, Ohta T, Takekawa Y, et al. (1997) Mitochondrial neurogasrtointestinal encephalomyopathy presenting with protein-losing gastroenteropathy and serum copper deficiency: a case report. Rinsho Shinkeigaku 37:917–922
Kocaefe YC, Erdem S, Özgüç M, et al. (2003) Four novel thymidine phosphorylase gene mutations in mitochondrial neurogastrointestinal encephalomyopathy syndrome (MNGIE) patients. Eur J Hum Genet 11:102–104
Chaury F, Fleury M, Tranchant C (2002) Cas clinique. Troubles digestifs révélateurs d’une cytopathie mitochondriale: le syndrome MNGIE. Rev Neurol (Paris) 158:111–113
Rafai MA, Laforet P, El Moutawakil B, et al. (2004) Mitochondriopathie type MNGIE «mitochondrial neuro-gastro-intestinal encephalopathy» [abstract]. Rev Neurol (Paris) 160(Spl.3):3S117
Martin MA, Blazquez A, Marti R, et al. (2004) Lack of gastrointestinal symptoms in a 60-year-old patient with MNGIE. Neurology 63:1536–7
Szigeti K, Wong LJ, Perng CL, et al. (2004) MNGIE with lack of skeletal muscle involvement and a novel TP splice site mutation. J Med Genet 41:125–9
Peker S, Pamir MN (2005) Trigeminal nevralgia in a patient with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). J Clin Neurosci 12:172–174
Gamez J, Lara MC, Mearin F, et al. (2005) A novel thymidine phosphorylase mutation in a Spanish MNGIE patient. J Neurol Sci 228:35–39
Slama A, Lacroix C, Plante-Bordeneuve V, et al. (2005) Thymidine phosphorylase gene mutations in patients with mitochondrial neurogastrointestinal encephalomyopathy syndrome. Mol Genet Metab 84:326–31
Martí R, Verschuuren JJ, Buchman A, et al. (2005) Late-onset MNGIE due to partial loss of thymidine phosphorylase activity. Ann Neurol 58:649–652
Blazquez A, Martín MA, Lara MC, et al. (2005) Increased muscle nucleoside levels associated with a novel frameshift mutation in the thymidine phosphorylase gene in a Spanish patient with MNGIE. Neuromuscul Disord 15:775–778
Hirano M, Garcia-de-Yebenes J, Jones AC, et al. (1998) Mitochondrial Neurogastrointestinal Encephalomyopathy Syndrome Maps to Chromosome 22q13.32-qter. Am J Hum Genet 63:526–533
Nishino I, Spinazzola A, Hirano M (1999) Thymidine Phosphorylase Gene Mutations in MNGIE, a Human Mitochondrial Disorder. Science 283:689–692
Hirano M, Martí R, Casali C, et al. (2006) Allogenic stem cell transplantation corrects biochemical derangements in MNGIE. Neurology 67:1458–1460
Bedlack RS, Vu T, Hammans S, et al. (2004) MNGIE neuropathy: Five cases mimicking chronic inflammatory demyelinating polyneuropathy. Muscle Nerve 29:364–368
Slotboom J, Boesch C, Kreis R (1998) Versatile Frequency Domain Fitting using Time Domain Models and Prior Knowledge. Magn Reson Med 39:899–911
Simmonds HA, Duley JA, Davies PM (1990) Analysis of purines and pyrimidines in blood, urine, and other physiological fluids. Chapter 25 In: Hommes FA, (Eds.), Techniques in diagnostic human biochemical genetics. A laboratory manual. John Wiley & Sons, 397–424
Rowland LP (1992) Progressive external ophthalmoplegia. In: Vinkens PJ, Bruyn GW, Klawans HL, (eds.) Handbook of clinical neurology. Elsevier, Amsterdam 287–329
Spinazzola A, Martí R, Nishino I, et al. (2002) Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem 277:4128–4133
Martí R, Nishigaki Y, Hirano M (2003) Elevated plasma deoxyuridine in patients with thymidine phosphorylase deficiency. Biochem Biophys Res Comm 303:14–18
Ferraro P, Pontarin G, Crocco L, et al. (2005) Mitochondrial deoxynucleotide pools in quiescent fibroblasts: a possible model for mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). J Biol Chem 280:24472–80
Hirano M, Nishigaki Y, Martí R (2004) Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): A disease of two genomes. Neurologist 10:8–17
Hirano M, Lagier-Tourenne C, Valentino ML, et al. (2005) Thymidine phosphorylase mutations cause instability of mitochondrial DNA. Gene 354:152–6
Martí R, Spinazzola A, Tadesse S, et al. (2004) Definitive diagnosis of mitochondrial neurogastrointestinal encephalomyopathy by biochemical assays. Clin Chem 50:120–124
Fairbanks LD, Marinaki AM, Carrey EA, et al. (2002) Deoxyuridine accumulation in urine in thymidine phosphorylase deficiency (MNGIE). J Inherit Metab Dis 25:603–604
Pfefferbaum A, Adalsteinsson E, Spielman D, et al. (1999) In vivo spectroscopic quantification of the N-acetyl moiety, creatine, and choline from large volumes of brain grey and white matter: Effects of normal aging. Magn Reson Med 41:276–284
Lundbom N, Barnett A, Bonavita S, et al. (1999) MR image segmentation and tissue metabolite contrast in 1H spectroscopic imaging of normal and aging brain. Magn Reson Med 41:841–845
Simmons ML, Frondoza CG, Coyle JT (1991) Immunocytochemical localization of N-acetyl-aspartate with monoclonal antibodies. Neuroscience 45:37–45
Miller BL, Chang L, Booth R, et al. (1996) In Vivo 1H MRS choline: correlation with in vitro chemistry/histology. Life Sci 58:1929–1935
Acknowledgements
We thank Dr. M. Hirano, Columbia University, New York, for the assessment of the leucocyte TP activity in the index patient. We also thank Dr. P. Voore, Toronto, Canada, for critical reading of this manuscript, Dr. JM Nuoffer, University Hospital Berne, Switzerland, for clinical advice, and J. Slotboom, PhD, for the development and implementation of the MRS-quantification technique and his assistance in acquiring the MR spectra for this study. This work was supported by grant No 81BE-67704 of the Swiss National Science Foundation and the Swiss Parkinson’s Disease Association.
Author information
Authors and Affiliations
Corresponding author
Additional information
Received in revised form: 26 March 2006
Rights and permissions
About this article
Cite this article
Schüpbach, W., Vadday, K.M., Schaller, A. et al. Mitochondrial neurogastrointestinal encephalomyopathy in three siblings. J Neurol 254, 146–153 (2007). https://doi.org/10.1007/s00415-006-0255-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-006-0255-3