
Abstract
Purpose
Liposomal cisplatin was developed to reduce the systemic toxicity of cisplatin, particularly the nephrotoxicity, and it has been used in combination with other agents in pancreatic and head and neck cancers and non-small-cell lung cancer (NSCLC). Our objective was to compare the effectiveness of lipoplatin combined with paclitaxel versus cisplatin with paclitaxel in advanced non-squamous NSCLC.
Methods
During 2007–2010, 202 patients with non-squamous NSCLC (stage IIIB and IV) were recruited from the two participating institutions and divided into two arms: Arm A was treated with liposomal cisplatin 200 mg/m2 combined with paclitaxel 135 mg/m2 and Arm B with cisplatin 75 mg/m2 in combination with paclitaxel 135 mg/m2, repeated every 2 weeks. The number of cycles administered was 632 (Arm A) and 640 (Arm B), totaling 1,272.
Results
A partial response was achieved by 59.22% of Arm A patients versus 42.42% of Arm B, and the difference was statistically significant (P 0.036). The median survival time in months was 10 for Arm A and 8 for Arm B (P 0.1551). After 18 months, the number of surviving patients was double for Arm A versus Arm B.
Conclusion
Liposomal cisplatin in combination with paclitaxel produces a statistically significantly higher response rate than cisplatin combined with paclitaxel in non-squamous NSCLC.
Similar content being viewed by others


Introduction
Over the last three decades, cisplatin (CDDP) has been one of the most effective cytotoxic drugs. Carcinomas of the head and neck, bladder, testicles, ovaries, esophagus, and lung are the most common malignancies that are sensitive to cisplatin when combined with a second or third cytotoxic agent [1–5]. Cisplatin has provided a survival advantage and a higher response rate, but its major problem has been toxicity, particularly nephrotoxicity [6, 7]. Over the years, attempts were made to find a substitute for cisplatin, mainly in the use of its analogue, carboplatin [5, 8]. Other cytotoxic agents such as taxanes (paclitaxel, docetaxel), gemcitabine, vinorelbine, pemetrexed, and irinotecan have also been used as substitutes [9–11]. However, none of these agents has proved to be superior to CDDP in effectiveness. Liposomal cisplatin (Lipoplatin, Regulon Inc., Mountain View, California) is another agent whose use over the last few years has been ongoing in trials. Lipoplatin is a new liposomal formulation formed from cisplatin and liposomes composed of dipalmitoyl phosphatidyl glycerol (DPPG), soyphosphatidyl choline (SPC-3), cholesterol, and methoxypolythylene glycol-distearoyl phosphatidylethanolamine (m-PEG 2000-DSPE). It was developed to reduce the systemic toxicity of cisplatin while attempting to improve the targeting of the drug to the primary tumor and to metastases by enhancing the half-life circulation time in body fluids and tissues. Preclinical studies have shown lipoplatin’s lower toxicity in rats, in comparison with cisplatin [12, 13]. Two phase I studies have tested lipoplatin’s pharmacokinetic profile and adverse reactions [14] and preferential tumor uptake in human studies [15]. Lipoplatin is administered in 1L 5% dextrose for an 8-hour infusion. The highest plasma concentration was defined at 6 h, and the platinum plasma levels were completely excreted after 4–7 days, the difference depending on the dosage of lipoplatin [14]. In a phase I–II trial, lipoplatin was combined with gemcitabine in patients with advanced pancreatic cancer; doses higher than 100–150 mg/m2 were well tolerated, and the only adverse reaction was grade 1–2 myelotoxicity [16]. Other trials have shown low or negligible nephrotoxicity [14, 17]. As monotherapy, lipoplatin is tolerated at a level of 350 mg/m2 [18]. Lipoplatin treatment of non-small-cell lung cancer (NSCLC) was tested in two randomized trials, a phase II and a phase III [19, 20]. It was found that the combination of lipoplatin with gemcitabine or with paclitaxel showed effectiveness similar to cisplatin plus gemcitabine or paclitaxel, but that there was much less toxicity with the lipoplatin combination. We also observed that there was a difference in the response rate of patients with non-squamous NSCLC, and this was an indication to run the present trial.
The aim of the present study was to evaluate the comparison between lipoplatin and cisplatin combinations with respect to response rate and median and overall survival in patients with non-squamous NSCLC.
Materials and methods
Patients’ eligibility
Eligibility for the study required histologically or cytologically confirmed NSCLC patients who were chemotherapy- and radiotherapy-naïve, classified as stage IIIB and IV and considered inoperable. Patients who had bidimensionally measurable disease on physical examination, X-rays, computed tomography (CT), WHO performance status (PS) of 0–2, expected survival ≥12 weeks, adequate bone marrow reserves (leukocyte count ≥3,500 μl−1, platelet count ≥100,000 μl−1, and hemoglobin ≥10 g μl−1), adequate renal function (serum creatinine ≤1.5 mg dl−1) and liver function (serum bilirubin not more than 1.5 mg dl−1 and serum transaminases not more than three times the upper limit of normal or not more than five times the upper limit of normal in cases of liver metastases), and age at least 18 years were eligible. In cases of central nervous system (CNS) involvement, patients were excluded unless they were asymptomatic. Patients with a second malignancy were also excluded. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines [21] and was approved by both participating hospital institutional ethics review boards. All patients gave their informed consent before entering the study.
Study design
The study was designed as a double-center phase III randomized trial. It was powered to detect a difference in the response rate and survival between the two arms. The sample was initially planned to include 200 patients (100 in each arm) with an increase in the number of patients if a statistical difference of 5% between the two arms, with regard to the response rate and to the median survival, was not reached. The randomization was performed centrally, and patients were stratified by three prognostic variables: disease stage (locally advanced versus metastatic disease), WHO PS of 0–2, and investigational site.
Statistical design
Patients were randomly assigned to the two treatment arms: Arm A was treated with lipoplatin plus paclitaxel and Arm B, cisplatin plus paclitaxel. Randomization was performed according to the method of random permuted blocks within strata. The stratification factor comprised disease stages IIIB and IV. Dynamic balancing by center was also performed. For time to disease progression and overall survival, the Kaplan–Meier method was used to estimate survival distribution and the log-rank test for the comparison of the treatment arms.
The response rates of the two treatment arms were calculated by the x2 test or Fisher’s exact test, when appropriate. The Mann–Whitney U test was used for toxicity-grade comparisons. All tests were two-sided. A P value less than 0.05 (Pearson’s chi-square test) was considered significant. The duration of response was calculated from the day of the first demonstration of response until PD. Progression-free survival (PFS) was calculated from the day of entry into the study until documented PD. Overall survival (OS) was calculated from the day of enrollment until death, or to the end of the study.
Treatment plan
Patients were randomly assigned to Arm A or Arm B. Arm A patients were to be treated with lipoplatin 200 mg/m2 in combination with paclitaxel 135 mg/m2. Lipoplatin was infused in 1L 5% dextrose for 8 h, without an extra infusion for hydration. Paclitaxel, given before lipoplatin, was infused for 3 h. Arm B patients were also given paclitaxel 135 mg/m2 for 3 h and cisplatin 75 mg/m2 in 250 ml normal saline solution accompanied by 1L 5% dextrose and 1L electrolyte. Premedication included ondansetron 8 mg intravenously (IV), dexamethasone 8 mg IV, and diphenyldramine 50 mg IV with modified timing 1 h before the beginning of treatment and repeated 4 and 8 h thereafter. Treatment of both arms was repeated every 2 weeks; this every 2-week treatment has been tested in other trials [22–24]. Nine cycles were planned. In repeating the treatment every 2 weeks, the dosages of cisplatin and paclitaxel were reduced to 75 mg/m2 instead of 100 mg/m2 for the former and 135 mg/m2 instead of 175 mg/m2 for the latter. Patients who responded to treatment continued to the end of the planned number of courses. Course delays of 1 week were permitted for recovery from adverse reactions. Concomitant supportive therapies, such as granulocyte colony-stimulating factors or blood transfusions, antibiotics and erythropoietic agents were allowed according to the ASCO guidelines [25].
Baseline and treatment assessment/evaluation
Before study entry, all patients underwent the following evaluations: medical history, physical examination, tumor measurement or evaluation, WHO PS, ECG, full blood count, liver and kidney function tests, and urinalysis. Staging was determined by chest and abdominal computed tomography, bone scan, and occasionally magnetic resonance imaging. Blood count, blood urea, and serum creatinine were measured before each treatment administration and 7 days after each course. During the treatment period, radiologic tests were conducted after four courses, at the end of the study and after any course if the clinical signs were indicative of disease progression. Disease status was assessed according to the response evaluation criteria in solid tumors. Randomly assigned patients who met the eligibility criteria were assessable for tumor response and duration of response. All patients in both arms who received at least one course of treatment were considered assessable for safety. Patients were assessed for toxicity according to the National Cancer Institute Common Toxicity Criteria, version 2.0. A complete response (CR) was considered to be the disappearance of all measurable disease confirmed at 4 weeks at the earliest; a partial response (PR), a 30% decrease in all measurable disease, also confirmed at 4 weeks at the earliest. In stable disease (SD), neither PR nor the progressive disease (PD) criteria were met; PD was considered to be a 20% increase in tumor burden and no CR, PR, or SD documented before increased disease [26]. A two-step deterioration in PS, a more than 10% loss of pretreatment weight, or increasing symptoms did not by themselves constitute progression of the disease; however, the appearance of these complaints was followed by a new evaluation of the extent of the disease. All responses had to be maintained for at least 4 weeks and be confirmed by an independent panel of radiologists and oncologists.
Results
From September 2007 until February 2010, 202 patients were enrolled in this double-center trial. All 202 patients (103 in Arm A and 99 in Arm B) were evaluable for response rate, survival, and toxicity. The patients’ characteristics are shown in Table 1; gender, age, PS, and histological or cytological examination are presented for both arms. The two arms of the study were well balanced with respect to the total number of patients and the aforementioned parameters.
Response evaluation
The response rate is shown in Table 2. No complete response was achieved in either of the two arms. In Arm A, partial remission was determined in 61/103 (59.22%) patients, stable disease in 35 (33.98%), and disease progression in 7 (6.80%). In Arm B, 42/99 (42.42%) patients achieved partial remission, 43 (43.43%) stable disease, and disease progression was determined in 14 (14.14%). There was a statistically significant difference in the response rate between the two arms in favor of Arm A (P 0.036, Table 2). No statistically significant difference with regard to age, gender, and tumor differentiation per arm was determined. The median duration of response for Arm A was 7 months (95% CI 4.6–9.4) and for Arm B, 6 months (95% CI 4.2–7.8). After the 10th month, there was a superiority in the survival rate in Arm A where 30% of the patients were without disease progression whereas in Arm B this was 16%.
Survival time to event
The median follow-up time was 18 months. The study ended in February 2010; by the end of the trial, there were 32 patients alive, 21 from Arm A (28.39%) and 11 from Arm B (11.11%).
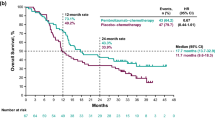
The median survival time for Arm A patients was 10 months (CI 95% 6.6–13.4) and for Arm B, 8 months (95% CI 5.4–10.6) (Fig. 1). No statistically significant difference was determined between the two arms (log-rank test P value 0.1551, Table 3). According to the statistics, the cumulative survival of 20 months in Arm A was 0.23 and in Arm B, 0.16.

Kaplan–Meier median survival time
Toxicity
Adverse reactions were mainly observed in Arm B patients treated with cisplatin–paclitaxel. In both arms, peripheral neuropathy was observed; it was more common in Arm B, but not significant. A statistically significant difference was detected in nausea/vomiting, asthenia, and particularly in nephrotoxicity (P < 0.001). With regard to these three side effects, less toxicity was observed in Arm A patients, treated with liposomal cisplatin and paclitaxel. A detailed toxicity analysis is reported in a previous trial [20]. There were no deaths owing to treatment-related toxicity.
Discussion
In a previous trial, liposomal cisplatin was shown to be less toxic than cisplatin when both were administered in combination with paclitaxel [20]. The two main histological types of NSCLC are squamous cell carcinoma and adenocarcinoma. It is possible that the different histological types of NSCLC do not have the same sensitivity to chemotherapy. Past trials have pointed out that the squamous cell type of lung cancer was more sensitive to chemotherapy than the adenocarcinoma cell type, with agents including cisplatin. For quite a number of years, adenocarcinoma of the lungs was considered to be rather insensitive to chemotherapy [9, 27, 28]. Past trials testing chemotherapy for lung cancers did not classify the histological subtypes, and all lung carcinomas were categorized as NSCLC for the treatment’s outcome [29–32]. Such indications from previous trials led us to run the present study where only patients with non-squamous NSCLC were recruited; one group was treated with liposomal cisplatin and the other with cisplatin, both combined with paclitaxel.
The statistically significant higher response rate of the patients who received liposomal cisplatin versus cisplatin in this study indicates that one should histologically subdivide the types of lung cancer by testing the response magnitude of different cytotoxic combinations. Parameters that are known and of importance in predicting the response and survival of patients with NSCLC may be tumor differentiation, disease stage and the site of the metastasis. On the basis of published data, certain trials have selected carboplatin as the platinum analogue of choice for first-line treatment of patients with metastatic NSCLC to avoid nephrotoxicity [33]. Two phase III trials comparing carboplatin plus paclitaxel with cisplatin-based combinations demonstrated similar efficacy, but lower rates of nausea, leukopenia, and nephrotoxicity with the use of carboplatin [9, 34].
Further clinical investigations comparing cisplatin versus carboplatin-based chemotherapy in the first-line treatment of advanced NSCLC are described in an individual patient data meta-analysis. In nine trials, which included 2,968 patients, the response rate was higher for patients treated with cisplatin than for patients treated with carboplatin (30% vs. 24%, respectively, overall survival P < 0.001). Carboplatin was associated with a statistically non-significant increase in the hazard ratio, versus treatment with cisplatin. These authors concluded that the patient meta-analysis suggests that cisplatin-based chemotherapy is slightly superior to carboplatin-based chemotherapy in terms of response rate and in certain subgroups [35]. Another comparison of chemotherapeutical agents in NSCLC histological groups showed that pemetrexed was more effective in adenocarcinoma and large-cell lung cancer than in squamous cell carcinoma [36]. This latter study was a retrospective analysis of a phase III trial comparing pemetrexed versus docetaxel in a second-line setting: the results indicated that patients treated with docetaxel had a statistically significant better survival rate than those treated with pemetrexed in the squamous cell subgroup, whereas pemetrexed rendered a statistically significant better survival rate in the non-squamous subgroup.
These data plus the outcome of our trial suggest that treatment selection of certain cytotoxic agents should be defined by the subtypes of NSCLC, thus resulting in a better response rate and survival.
Liposomal cisplatin combined with paclitaxel showed a statistically significant higher response rate than cisplatin combined with paclitaxel in the treatment of non-squamous carcinomas of the lung.
References
Livingston RB (1977) Combination of chemotherapy of bronchogenic carcinoma, I. Non-oat cells. Cancer Treat Rev 4:153–165
Al-Sarraf M (1987) Chemotherapeutic management of head and neck cancer. Cancer Metastasis Rev 6:181–198
Sternberg CN, Yagoda A, Scher HI, Watson RC, Geller N, Herr HW, Tannock I, Gespedarowitz M, Connelly J, Jewett M (1989) Methotrexate, vinblastine, doxorubicin, and cisplatin for advanced transitional cell carcinoma of the urothelium, efficacy and patterns of response and relapse. Cancer 64:2448–2458
Oliver T, Mead G (1993) Testicular cancer. Curr Opin Oncol 5:559–567
Taylor AE, Wiltshaw E, Gore ME, Fryatt I, Fischer C (1994) Long-term follow-up of the first randomized study of cisplatin versus carboplatin for advanced epithelial ovarian cancer. J Clin Oncol 12:2066–2070
Humes HD (1999) Insights into ototoxicity, analogies to nephrotoxicity. Ann NY Acad Sci 884:15–18
Arany I, Safirstein RL (2003) Cisplatin nephrotoxicity. Semin Nephrol 23:460–464
Caraceni A, Martini C, Spatti G, Thomas A, Onofrj M (1997) Recovering optic neuritis during systemic cisplatin and carboplatin chemotherapy. Acta Neurol Scand 96:260–261
Non-Small Cell Lung Cancer Collaborative Group (1995) Chemotherapy in non-small-cell lung cancer: a meta-analysis using updated data on individual patients from 52 randomised clinical trials. BMJ 311:899–909
Stathopoulos GP, Rigatos S, Malamos NA (1999) Paclitaxel combined with cisplatin as second-line treatment in patients with advanced non-small-cell lung cancers refractory to cisplatin. Oncol Rep 6:797–800
Piccart MJ, Bertelsen K, James K, Cassidy J, Mangioni C, Simousen E, James K, Kaye S, Vergote J, Blom R, Grimshaw R, Atkinson R, Swenerton K, Trope C (2000) Randomized intergroup trial of cisplatin-paclitaxel versus cisplatin-cyclophosfamide in women with advanced epithelial ovarian cancer: three-year results. J Natl Cancer Inst 92:699–708
Boulikas T (2004) Low toxicity and anticancer activity of a novel liposomal cisplatin (lipoplatin) in mouse xenografts. Oncol Rep 12:3–12
Devarajan P, Tarabishi R, Mishra J, Ma Q, Kourvetaris A, Vougiouka M, Boulikas T (2004) Low renal toxicity of lipoplatin compared to cisplatin in animals. Anticancer Res 24:2193–2200
Stathopoulos GP, Boulikas T, Vougiouka M, Delikonstantinos G, Rigatos S, Darli E, Viliotou V, Stathopoulos JG (2005) Pharmacokinetics and adverse reactions of a new liposomal cisplatin (lipoplatin), phase I study. Oncol Rep 13:589–595
Boulikas T, Stathopoulos GP, Volakakis N, Vougiouka M (2005) Systemic lipoplatin infusion results in preferential tumor uptake in human studies. Anticancer Res 25:3031–3040
Stathopoulos GP, Boulikas T, Vougiouka M, Rigatos SK, Stathopoulos JG (2006) Liposomal cisplatin combined with gemcitabine in pretreated advanced pancreatic cancer patients: a phase I–II study. Oncol Rep 15:1201–1204
Arienti Ch, Tesei A, Ravaioli A, Ratta M, Carloni S, Mangianti S, Ulivi P, Nicoletti S, Amadori D, Zoli W (2008) Activity of lipoplatin in tumor and in normal cells in vitro. Anticancer Drugs 19:983–990
Stathopoulos GP, Rigatos SK, Stathopoulos J (2010) Liposomal cisplatin-dose escalation for determining the maximum tolerated dose and dose-limiting toxicity: a phase I study. Anticancer Res 30:1317–1322
Mylonakis N, Athanasiou A, Ziras N, Angel J, Rapti A, Lampaki S, Politis N, Karanikas C, Kosmas C (2010) Phase II study of liposomal cisplatin (Lipoplatin™) plus gemcitabine versus cisplatin plus gemcitabine as first line treatment in inoperable (stage IIIb/IV) non-small-cell lung cancer. Lung Cancer 68:240–247
Stathopoulos GP, Antoniou D, Dimitroulis J, Michalopoulou P, Bastas A, Marosis K, Stathopoulos J, Provata A, Yiamboudakis P, Veldekis D, Lolis N, Georgatou N, Toubis M, Pappas Ch, Tsoukalas G (2010) Liposomal cisplatin combined with paclitaxel versus cisplatin and paclitaxel in non-small cell lung cancer: a randomized phase III multicenter trial. Ann Oncol 21:2227–2232
ICH Efficacy Guidelines EG (R1): Good Clinical Practice consolidated guidelines, http://ich.org/cache/compo/475-175.html
Chen CH, Lin CH, Kao KC, Chauge JW, Tsap TC (2007) Phase II study of a biweekly regimen of vinorelbine and cisplatin in advanced non-small-cell lung cancer. Chang Cung Med 30:249–255
Tas F, Guney N, Derin D, Camlica H, Aydiner A, Topuz E (2008) Bi-weekly administration of gemcitabine and cisplatin chemotherapy in patients with anthracycline and taxanes-pretreated metastatic breast cancer. Invest New Drugs 26:363–368
Heinrish S, Pestallozi BC, Schafer M, Weber A, Bauerfeind P, Knuth A, Clavien PA (2008) Prospective phase II trial of neoadjuvant chemotherapy with gemcitabine and cisplatin for resectable adenocarcinoma of the pancreatic head. J Clin Oncol 20:2526–2531
Arbuck SG, Ivy SP, Setser A. The revised common toxicity criteria: version 20, http://ctep.info.nih.gov
Therasse P, Arbuck SG, Eisenhower EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, Van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors: European Organization for Research and Treatment of Cancer. National Cancer Institute of the United States. J Natl Cancer Inst 92:205–216
Rapp E, Pater JL, Willan A, Cormier Y, Murray N, Evans WK, MacKillop WJ, Ward GK, O’Sullivan B (1988) Chemotherapy can prolong survival in patients with advanced non-small-cell lung cancer, report of a Canadian multicenter randomized trial. J Clin Oncol 6:633–641
Van Zandwijk N, Smit EF, Kramer GWP, Sihramel F, Gans F, Festen J, Huisman C, Giaccone G, Van Groeningen CJ (2000) Gemcitabine and cisplatin as induction regimen for patients with biopsy-proven stage IIIA N2 non-small-cell lung cancer: a phase II study of the European Organization for Research and Treatment of Cancer, Lung Cancer Cooperative Group (EORTC). J Clin Oncol 18:2658–2664
Rosell R, Gomez-Codina J, Camps C, Maestre J, Padille J, Canto A, Mate JL, Li S, Roig J, Olazabal A (1994) A randomized trial comparing preoperative chemotherapy plus surgery with surgery alone in patients with non-small-cell lung cancer. NEJM 330:153–158
Ruckdeschel JC, Finkelstein DM, Mason BA, Creech RH (1985) Chemotherapy for metastatic non-small cell broncohogenic carcinoma, EST 2575, generation V-a randomized comparison of four cisplatin containing regimens. J Clin Oncol 3:72–79
Souquet PJ, Chauvin F, Boissel JP, Cellerino R, Cormier Y, Ganz PA, Clarke SJ, Boyer MJ (1993) Polychemotherapy in advanced non-small cell lung cancer: a meta-analysis. Lancet 342:19–21
Giaccone G (2004) Twenty-five years of treating advanced NSCLC: what have we achieved? Ann Oncol 15:Siv81–Siv83
Azzoli CG, Kris MG, Pfister DG (2007) Cisplatin versus carboplatin for patients with metastatic non-small-cell lung cancer, an old rivalry renewed. JNCI 99(11):828–829
Jemal A, Murray T, Ward E, Samuels A, Tiwari R, Ghafoor A, Ma Y, Peng J, Liu W, Zhang P, Huang L, Gao B, Shen T, Zhou Y, Chen H, Chu (2005) Cancer statistics, 2005, CA. Cancer J Clin 55:10–30
Ardizzoni A, Boni L, Tiseo M, Fosella FV, Schiller JH, Paesmaus M, Randosavljevic D, Paccagnella A, Zatloukal P, Mazzanti P, Bisset D, Rossel R (2007) Cisplatin versus carboplatin based chemotherapy in first-line treatment of advanced non-small cell lung cancer: an individual patient data meta-analysis. JNCI 11:847–857
Peterson P, Park K, Fosella FV, Gatzemeier U, John W, Scallioti GV, Ciuleanu TE, Brodowicz T (2005) 12th world conference on lung cancer. In: Is pemetrexed more effective in adenocarcinoma and large cell lung cancer than in squamous cell carcinoma? A retrospective analysis of a phase III trial of pemetrexed versus docetaxel in previously treated patients with advanced non-small cell lung cancer (NSCLC). 12th World Conference on Lung Cancer, pp P2-328 (abstract)
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Stathopoulos, G.P., Antoniou, D., Dimitroulis, J. et al. Comparison of liposomal cisplatin versus cisplatin in non-squamous cell non-small-cell lung cancer. Cancer Chemother Pharmacol 68, 945–950 (2011). https://doi.org/10.1007/s00280-011-1572-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-011-1572-5