
Abstract
Hemp cultivation is living a period of renewed interest worldwide after long years of opposition and abandonment. The European Union (EU) allows and subsidizes the growing of fiber and oilseed cultivars of Cannabis sativa L. with respect to the THC content limit of 0.2%. The EU method for the quantitative determination of Δ9-tetrahydrocannabinol (THC) content in hemp varieties provides to apply a tolerance of 0.03 g of THC per 100 g of sample concerning compliance assessment to that limit. However, the method does not report any precision data, especially useful as a function of THC content to evaluate measurement uncertainty and therefore to establish the conformity of hemp at different THC legal limits. Measurement uncertainty of the method by both bottom-up and top-down approach, besides repeatability and reproducibility, was investigated and estimated in the THC concentration range 0.2–1.0%, which includes the different legal limits set out for hemp around the world. We proposed Decision Rules for conformity of hemp showing that a non-compliant declaration beyond reasonable doubt should be stated when the THC content, as a mean result on a duplicate analysis, exceeds the limit by at least 11–15%, depending on THC limit. We highlighted other issues concerning practical aspects of hemp analysis, from sampling to evaluation of results, as well as the need to carry out collaborative studies on the EU method.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cannabis sativa L. is the world’s most recognizable, notorious, and controversial plant known since the ancient times for its medicinal and textile uses, an emblematic example of a multi-purpose crop [1, 2]. It is also by far the most widely cultivated, trafficked, and abused illicit drug [3].
The name “hemp” or “industrial hemp” designates fiber and oilseed cultivars of C. sativa with very limited content of Delta-9-tetrahydrocannabinol (Δ9-THC, or simply THC). Conversely, “marijuana” is the name used for the drug kind of plant, containing a high level of THC. THC and CBD (cannabidiol) are the plant phytocannabinoids of most importance. THC is the principal intoxicant and psychotropic constituent, while CBD, devoid of psychotropic effects and known to possess several pharmacological properties, is instead the principal cannabinoid of hemp [4]. These compounds, as other cannabinoids, exist in the fresh plant mostly in the form of carboxylic acids, THCA and CBDA, possessing several pharmacological properties but no psychotropic activity [5]. These acids undergo decarboxylation into their neutral counterparts under the influence of light, time (such as prolonged storage), alkaline conditions, or high temperature (smoked or cooked marijuana) following the reaction shown in Fig. 1.

The decarboxylation reaction of THCA to THC
Marijuana has been considered a leading drug of abuse and has been seriously criminalized, with enormous law enforcement costs and social upheaval [1]. It is currently included in Schedule I of the United Nation (UN) Single Convention on Narcotics Drugs (1961) [6] and only recently it was removed from Schedule IV, the most restrictive [7]. In the last decade, decriminalization of Cannabis for industrial and medicinal uses and even recreational marijuana has occurred, or it is occurring in many jurisdictions as the result of sociological, philosophical, political, and legal developments [8]. A limit of 0.3% of THC content (on a dry inflorescences weight basis) was established by Small et al. (1976) [9] and adopted in many countries as a criterion to distinguish cultivars that can be legally cultivated under license from those considered to have a too high drug potential [1]. Some jurisdictions have increased this limit for legal cultivars up to 1.0% [1, 10, 11].
In Europe, the THC limit value for industrial hemp was first set at 0.5% in 1984, then trimmed to 0.3% in 1987, and further lowered to 0.2% in 1999 to prevent the cultivation of illicit drug–type Cannabis in hemp fields [12]. The EU subsidies to hemp cultivation are granted upon the use of certified seeds from the varieties listed in the “Common Catalogue of Varieties of Agricultural Plant Species” [13], provided that THC content does not exceed 0.2% [14]. Hemp imported in the EU must meet the same limit [15].
In Italy the Law n. 242/2016 [16], laying down rules for support and promotion of the hemp cultivation, stated that farmers were not liable to the Italian narcotics law [17] when hemp THC content, higher than 0.2%, did not exceed the 0.6%. This led to the misinterpretation of the Law as a general liberalization of Cannabis derivatives having a THC content below 0.6%, the so-called Cannabis light. The consequent booming demand of C. light and the specialized stores opening (+200% from 2016) [18] quickly became an economic and social phenomenon, giving rise to many jurisdictional controversies and to several seizures. A note of the Italian Ministry of Interior stated, in 2018, that Cannabis inflorescences, plants, concentrates, essences, and resins on the market were considered narcotics when the THC content was higher than 0.5%, according to a judgment of the Italian Supreme Court of Cassation (1989) based on forensic toxicology studies, scientific literature, and court judgments [19]. In 2019, the Italian Supreme Court (plenary session) stated that the marketing of leaves, inflorescences, oil, and resin of Cannabis sativa L. was out of the scope of the Law n.242/2016 and then it is an offense under the Italian drug control law, “except if the products are in practice devoid of narcotic effects.”
Actually, the current legal framework in this field needs to be harmonized and better defined to avoid ambiguous interpretations and contradictory judgments. Anyway, Authorities and law enforcement agencies worldwide have in charge to analyze Cannabis derivatives verifying its compliance with different THC legal limits. As required by ISO/IEC 17025:2017, the applied decision rules must be clearly defined when reporting about compliance, and the knowledge of uncertainty associated with the measurements is essential to this purpose. Without this information, there is a risk of misinterpretation of results, incorrect prosecution in law, adverse health, or social consequences [20].
A widely adopted standard approach to uncertainty evaluation is the Guide to the Expression of Measurement Uncertainty (GUM) [21], using a bottom-up approach. On the other hand, it is possible to carry out collaborative studies on standard test methods, and measurement uncertainty evaluation is achieved using precision and trueness estimates (top-down approach) [22].
Literature reports several analytical methods for determining phytocannabinoids in Cannabis plants and derivatives, most of which based on gas chromatography coupled to flame ionization (GC-FID) or mass spectrometry (GC-MS) detector, or on high-performance liquid chromatography coupled to ultraviolet (HPLC-UV) or mass spectrometry (HPLC-MS) detector [5, 23,24,25,26,27,28,29,30]. Each method has inherent limitations and many are the pitfalls encountered in Cannabis analysis, from sampling to sample preparation and from cannabinoids extraction to instrumental analysis [31]. In particular, these issues may be highly critical in the analysis of samples with low content of THC.
The official method established by the European Commission employs a GC-FID analysis [32]. However, this method does not report any precision data, useful especially as a function of THC content to measurement uncertainty evaluation and therefore to establish the conformity of hemp at different THC legal limits. It reports applying a tolerance of 0.03 g of THC per 100 g of sample for compliance to the limit of 0.2% THC content.
Therefore, we considered it necessary to investigate and estimate the precision data of the EU method at different THC concentration levels, representing the THC legal limits set out for hemp around the world: 0.2%, 0.3%, 0.5%, 0.6%, and 1.0%. We verified the specificity of the method by GC-MS analysis and its trueness by comparing its data with those obtained by a GC-FID method validated and accredited at the Italian Customs and Monopolies Agency Laboratories (MAD) [33]. We evaluated the measurement uncertainty for each one of the abovementioned legal limits by both bottom-up and top-down approaches and decision rules for compliance assessment of hemp were proposed.
Materials and method
Reagents and solvents
Δ9-tetrahydrocannabinol (Δ9-THC, 1.0 mg/ml in methanol) and Δ9-tetrahydrocannabinoid acid A (THCA, 1.0 mg/ml in acetonitrile) were supplied by Cerilliant (Round Rock, Texas), squalane (analytical Internal Standard, IS, > 99%) was purchased from Merck (Darmstadt, Germany), and n-hexane (99%) was purchased from Carlo Erba (Milano, Italy). The dried hemp inflorescence samples were seized by Authorities or delivered in the analysis by growers and retailers.
Sample preparation
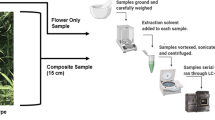
First, we verified the moisture content in the samples. Shortly, about 3 g of each sample was accurately weighed and further dried for 4 h at 103 °C in an oven [34], weighted again, and then discarded.
The samples, according to the EU method [32], were ground to a semi-fine powder (passing through a 1-mm mesh sieve), after removing stems and seeds over 2 mm in size. We placed 100 mg of the powdered sample in a centrifuge tube adding 5 ml of extraction solution containing the internal standard (IS) (35 mg of squalane per 100 ml hexane). The sample was placed in an ultrasound bath for 20 min, then centrifuged for 5 min at 1390 g. The supernatant, containing the THC, was removed and placed in a vial for GC analysis.
In particular, we selected, or prepared by properly mixing the available real ones, five samples at different concentration levels, ranging from about 0.1 to 1.0% m/m of THC.
Calibration curve
The calibration standard solutions were prepared by drying a proper volume of the THC standard solution in methanol under N2 flow and diluting it by the IS extraction solution. The calibration curve included 5 calibration levels, 0.02, 0.04, 0.12, 0.25, and 0.5 mg/ml of THC, corresponding to a THC amount in samples ranging from 0.1 to 2.5% m/m. In particular, the 0.04 and 0.5 mg/ml standard solutions were those required by the EU method. We injected 3 times each standard solution and plotted the THC and IS peaks area ratio vs their concentration ratio.
The calibration curve equation was calculated by the least squares regression and linearity evaluated by the coefficient of determination and normalized residuals. The limit of detection (LOD) and the limit of quantification (LOQ) were estimated on a 3:1 and a 10:1 signal-to-noise ratio, respectively.
GC-FID analysis
The GC-FID analyses according to the EU method were performed on a Shimadzu GC-2010 Plus SSL/FID with autosampler (Shimadzu Corporation, Kyoto, Japan), equipped with a Restek Rxi-5-ms fused silica capillary column, 30 m × 0.25 mm i.d., and 0.25-μm film thickness (cross-linked 5% diphenyl-95% dimethylpolysiloxane). The carrier gas was helium, at a flow rate of 1 ml/min. The injection volume was 1 μl with a 1:40 split ratio. The oven temperature was set to 260 °C × 10 min, then to 300 °C (20 °C/min) × 2 min. The injector and FID temperature was set at 300 °C, the latter fed by a flow of H2 (40 ml/min), air (400 ml/min), and N2 (30 ml/min) as make-up gas. Each single run lasted 14 min.
The THC amount (y) was calculated by the following formula:
where a and b are respectively the intercept and the slope of the linear regression equation, A is the chromatographic peak area, C is the concentration in mg/ml, Vsol is the extraction solution volume in ml, and ws is the sample weight in mg.
The analyses according to the MAD method [33] were performed on the same GC-FID instrument. This method differs from the EU one in the extraction solution (IS and solvent) and in the GC temperatures.
GC-MS analysis
The GC-MS analyses to identify the extracted compounds and to evaluate the method specificity were performed on a Thermo Focus GC/DSQ II with autosampler (Thermo Fisher Scientific, MA, USA). The column and the GC conditions were the same reported above for the EU GC-FID assay. The MS detection was performed by electron ionization (EI) at 70 eV, operating in full scan acquisition mode in the m/z range 40–450. The interface and ion source temperatures were set at 270 and 250 °C, respectively. The reference standard solutions were run under the same conditions and mass spectra matches were carried out using the National Institute of Standards and Technology (NIST) mass spectra database (version 2.2, 2014).
Statistical analysis
The precision, trueness, and uncertainty of the THC content measurement were determined by following international guidelines [35,36,37,38] and by applying standard statistical treatments to experimental data. The variability (repeatability) associated to the THC content was studied by the Shapiro-Wilk test to verify the normal distribution of data and by Dixon, Grubbs, and Huber test to remove the outlier data. We applied the t-test, the F-test, and the Hartley test to study the means and the variances, respectively.
The Horwitz ratio (HorRat), a performance parameter reflecting the acceptability of a chemical method of analysis with respect to precision, was applied to evaluate the within-laboratory variability [39].
We determined the measurement uncertainty, which characterizes the dispersion of the values reasonably attributed to the analytes, by both the bottom-up approach [21] and the top-down one [22].
Results and discussion
Specificity of the method
Some sample extracts underwent analysis by both GC-FID and GC-MS assays to ascertain the specificity of the EU method. The same profiles and similar peak retention times resulted from the two techniques. For the THC and the IS peaks, a resolution higher than 1.3 occurred in all samples. We obtained the identification of the different cannabinoids by comparing our retention time of peaks and MS fragmentation patterns with those of the reference standards (Figs. 2 and 3). The method, therefore, resulted specific.

GC-FID chromatogram of the extract from a hemp sample with low content of THC

GC-MS chromatogram and mass spectrum of the extract from a hemp sample with low content of THC
Calibration curve
The calibration curve equation for the GC analyses resulted y = 0.8216x − 0.0155, with R2 = 0.9995 and normalized residuals <2. The limit of detection (LOD) and quantification (LOQ) resulted equal to 0.003 mg/ml and 0.010 mg/ml, respectively. These values correspond to a THC content of 0.015% and 0.050% m/m, less than one tenth and equal to one fourth of the legal limit of 0.2%.
Precision
The five selected or prepared samples were analyzed by performing eleven independent replications, in order to assess the variability (repeatability) associated to the THC level, which was in the range 0.10–1.11% m/m of THC. The moisture content in all samples was in the range 8–13%, as required by the EU method.
The results of data statistical treatment are reported in Table 1. For each concentration level, we calculated:
-
The mean measured value of THC (%),
-
The repeatability (within-laboratory) standard deviation (sr),
-
The relative standard deviation (RSDr,%),
-
The predicted reproducibility (among-laboratory) standard deviation calculated by the Horwitz equation (σH(%m/m) = 2C0.8495), where C is the THC content as mass fraction m/m),
-
The predicted relative standard deviation (PRSDR = σH/THC,%),
-
The Horwitz ratio (HorRatr = RSDr/ PRSDR).
The Hartley test on variances allowed establishing that variability of the analysis was dependent from THC level, being:
where α = 0.05 (significance level); p = 5 (number of levels); υ = 10 (degrees of freedom).
In particular, the standard deviation sr showed a linear dependence from the THC % m/m in the investigated range, which equation was y = 0.035x + 0.0018 with an R2 = 0.9918.
This result confirmed that our choice to analyze just only the five samples corresponding to the legal limits set around the world led to statistically significant results.
The original Horwitz ratio, HorRatR (RSDR/PRSDR, where RSDR is the relative reproducibility standard deviation obtained by an inter-laboratory study), has empirical acceptable values in the range 0.5–2.0 [39]. To the best of our knowledge, collaborative trials on the UE method were not performed yet, nor were available precision data (sR) for the analysis of THC low-level contents.
Anyway, the Horwitz ratio can be also applied to within-laboratory precision (HorRatr), although with less reliability. Since the within-laboratory variability (sr) is typically one half to two thirds of the among-laboratory variability (sR) [39], the HorRatr acceptable range shall be 1/4–4/3. As shown in Table 1, at all analyzed THC levels, the required condition was satisfied.
Trueness
Certified reference materials (CRM) for hemp were not available and we evaluated the trueness of data by two assays.
We employed a THC standard solution at 0.1 mg/ml, an intermediate level of calibration, corresponding to the 0.5% amount in a sample. We injected this standard sample in triplicate and the recovery values resulted in the range 99.8–100.3%.
In parallel, we compared the results obtained on six replicates of a randomly selected real sample, analyzed by both the EU method and the MAD one. We applied the t-test and the F-test to the mean and variance values. It was noteworthy that the two GC-FID methods were perfectly comparable. In fact, the mean value was 0.28% for both methods and the standard deviation resulted 0.0099% for the EU method and 0.0072% for the MAD one.
Moreover, we decided to evaluate the THCA conversion and recovery values in our GC system. Indeed, THC is mostly present as a carboxylated form in fresh Cannabis plants as well as in samples dried at low temperature and not too old. The EU method, in fact, provides to dry fresh hemp samples below 70 °C, since at higher temperature (85–100 °C) the THC decomposition may occur. THCA decarboxylation reaction starts around 60–90 °C [40,41,42,43,44] and conversion to THC was never perfectly complete without loss or degradation of the starting material [45, 46].
As already reported by Dussy et al. [43], the THCA decarboxylation reaction occurring in a gas chromatograph does not have a fixed rate as it strictly depends on the liner geometry and injector port temperature. It was referred a thermal conversion of THCA of only 70% at the maximum.
Therefore, we employed a THCA standard solution (STDS) at three different concentrations, each one injected three times and results were quantified as THC. We reported the results in Table 2.
The THCA mean recovery value denoted a satisfactory performance of our GC system relating to the THCA conversion rate throughout the concentration range of our interest.
In any case, a correction of the results by THCA recovery is neither advisable nor is it effectively possible, since the actual THCA content in Cannabis products cannot be determined by a GC analysis without any prior derivatization. Nevertheless, THCA recovery and conversion rate determination are recommended to verify and ensure the own GC is suitable to perform the analysis [23].
In general, it is possible to affirm that Cannabis analysis by GC may cause a variable underestimation of total THC content.
Measurement uncertainty
The bottom-up approach
This methodology evaluates the measurement uncertainty focused on individual input quantities. In general, this approach may underestimate the measurement uncertainty, mainly because it can be difficult to identify and include all possible contributions.
Shortly, the combined standard uncertainty uc(y) associated to a result (y) is determined from the estimated standard deviation associated with each input xi, the standard uncertainty u (xi). Some of these inputs are evaluated from the statistical distribution of the results and are characterized by experimental standard deviations (type A evaluation). Other inputs, also characterized by standard deviations, are evaluated from assumed probability distributions based on experience or on other information (type B evaluation).
The relative combined standard uncertainty,\( {\dot{\ u}}_c(y) \), considering the Eq. (1), was calculated as:
by combining the contributions of repeatability (urep), a type A evaluation, with those of regression curve (ureg), THC certified reference material (uCRM(THC)), IS certified reference material (uCRM(IS)), sample weight (uws), and extraction solution volume (uVsol), representing type B evaluations.
In case the moisture content does not fall within the range provided by the method and a correction is therefore necessary, the related contribution should be included in the uncertainty budget as well.
The expressions employed to calculate each contribution value are provided in the Supplementary Information (ESM).
It is appropriate to underline that we used a modified version of the well-known formula for ureg calculation, specifically omitting the first term “1/m” (where m is the number of replicates) provided under the radical. Indeed, it is necessary to avoid double counting the precision contribution, already accounted in the uncertainty budget with the repeatability term \( {\dot{u}}_{rep} \) that incorporates all the individual sources of variability, including that relating to calibration, as well explained by Kadis [47].
For each legal limit in the range 0.2–1.0% of THC, the combined standard uncertainty, uc(y) = y\( {\dot{u}}_c(y) \), and the expanded uncertainty, U(y) = k uc (y), were calculated both for one (m = 1) and two (m = 2) determinations per test sample.
Expanded uncertainty was calculated using a coverage factor k = 2, as the effective degrees of freedom resulted νeff > 10, providing a level of confidence of approximately 95%.
In Table 3, we reported the values of the abovementioned inputs and the combined relative standard uncertainties, whereas in Table 4, the standard, expanded, and relative expanded uncertainty values.
The \( {\dot{u}}_{rep} \) and the \( {\dot{u}}_{reg} \) values depend on THC amount and were extrapolated by the respective correlation functions.
The correlation values and function of \( {\dot{u}}_{reg} \) with the THC content (both mg/ml and %) are reported in Table S1 and Fig. S1 in the ESM.
The calibration uncertainty ucal, given by the appropriate sum of ureg, uCRM(THC), and uCRM(IS), represented about 40% (60% for m = 2) of the global uncertainty at the 0.2% THC. It decreased to 10% (20% for m = 2) for the 1.0% limit. A graph (Fig. S2) showing the contribution of each input to the combined standard uncertainty at each legal limit was included in the ESM.
The top-down approach
The measurement uncertainty can be evaluated also by using the repeatability, reproducibility, and trueness data obtained by collaborative studies conducted in accordance with ISO 5725-2 [22, 48], following the principle that reproducibility standard deviation obtained on a collaborative study is a valid basis for measurement uncertainty evaluation.
According to ISO 21748/2017 [22], we calculated the standard uncertainty by the following equation:
Hence, the expanded uncertainty is calculated as U(y) = ku(y), where k = 2.
Since sR information from collaborative study on EU method are unavailable to date, we calculated a rough estimate of sR from sr, as:
(x = % THC content), considering that the “Horwitz ratio” (sR/sr) for analytical procedures is typically close to 2.0 and does not change significantly with the concentration of the analyte [49]. This calculation of sR was still within the acceptable range of the HorRatR parameter, being less than two.
In the case of one determination per test sample (m = 1), as required by procedure A of the EU method, the expanded uncertainty resulted from:
In the case of duplicate determinations (m = 2), procedure B, as it is required when the single result is above the allowed limit, the equation became:
In Table 5, we reported the standard, expanded, and relative expanded uncertainty values obtained by the top-down approach.
The top-down approach may not by itself identify where the major errors occur and the results depend on technical competence of the laboratory concerned.
Comparison of the uncertainties values
The bottom-up and top-down standard uncertainties were compared for each legal limit by the F-test and were considered significantly different at a 95% confidence level. For both approaches, the relation between repeatability and uncertainty, r (= 2\( \sqrt{2} \)sr) < 2 U, was verified.
We ascertained the linear correlation existing between the expanded uncertainties and the THC content, for both approaches, as shown in Fig. 4: the top-down uncertainty resulted about 1.5–2 times larger than the bottom-up one.

Linear correlation between top-down (TD) or bottom-up (BU) expanded uncertainties (for m = 1 and m = 2) and the THC content
In Table S2 in the ESM, all the ratios of the combined standard uncertainties for m = 1 and m = 2 were reported together with the predicted standard uncertainty.
We supposed that the bottom-up approach underestimated the measurement uncertainty in hemp analysis. Indeed, the bottom-up standard uncertainty resulted quite similar to the repeatability standard deviation sr, although also comparable to the predicted reproducibility standard deviation (σH) calculated by the Horwitz equation [39]. However, in relation to the latter aspect, it is possible to hypothesize a larger predicted variability: recent collaborative trials [49] showed a sR significantly higher than that one provided by Horwitz, resulting much closer to the top-down standard uncertainty obtained.
Hence, inter-laboratory studies on hemp samples with low THC content will be necessary to estimate the actual precision data of the EU method. The collaborative trials (CT) will be the most suitable for this purpose. The proficiency tests (PT) do not usually prescribe specific methods and the inter-method differences will increase dispersion and uncertainty associated to the results. Statistical treatments showed that the inter-laboratory standard deviation sR’ under PT conditions is higher than sR under CT ones, on average sR’ ≈ 1,5sR [49, 50].
The uncertainty may result even larger considering the issue of THCA conversion variability and other sources of uncertainty, not accounted even by a collaborative study, such as sampling, removing stems and seeds (sub-sampling), and drying and grinding of samples.
We considered the top-down uncertainty estimated in this work as the minimum one to be associated to the result and the compliance assessment of hemp was based on it.
Compliance assessment
The decision rules give a prescription for the acceptance or rejection of a product based on the measurement result, its uncertainty, and the specification limit or limits, taking into account the acceptable level of the probability of making a wrong decision [51,52,53].
We proposed a decision rule for non-compliance with a low probability of false rejection (high confidence of correct rejection) consistent with that one adopted by the EU method, allowing a tolerance beyond the admitted limit. Currently, a widely used decision rule implies non-compliance with an upper limit if the measured value exceeds the limit by the expanded uncertainty [51]. Anyway, the use of guard bands is preferred, as it can reduce the probability of making an incorrect conformance decision [52]. The rejection zone starts at the value of the specification limit L plus an amount g (guard band). The value of g depends upon the value of the uncertainty and the values resulting in greater than L + g have a probability of false rejection lower than the risk “α” [51], which typical value is 5%. The probability, P, that the value higher than L + g is actually greater than the limit L is at least 95%, i.e., “beyond reasonable doubt” [54, 55]. The size of the guard band was g = ku, where k = 1.65 for the decision based on one-tailed significance test at 95%; in other terms, g corresponded to about 0.83 U.
We considered two possibilities, calculating the uncertainty at the legal limit (uL) or at the measured value (uy). The latter gives a larger guard band, as u was proportional to y. Moreover, the uncertainty estimated for two determinations (m = 2) was taken into account since the THC measurement in a sample exceeding the limit must be repeated [21].
In Table 6, we reported for each legal limit, by using the top-down uncertainty (U = 0.1308 THC(%) + 0.0066, for m = 2), the maximum THC content (L + g) beyond which non-compliance of hemp samples should be declared.
The differences between the two approaches resulted minimal and measurable only for the higher limits. It is worth to note that at the 0.2% limit, the band guard coincides with the tolerance (0.03%) applied by the EU method, supporting the choice of the top-down uncertainty.
Conclusion
The precision data and measurement uncertainty of the EU method for THC determination in hemp, investigated in the range 0.2–1.0%, showed a linear dependence with THC content.
We evaluated measurement uncertainty, essential to define decision rules for compliance assessment, by both bottom-up and top-down approaches and the latter resulted more suitable for the purpose.
We proposed decision rules for each THC legal limit, which resulted consistent with the strategy adopted by the EU method to determine the tolerance for the 0.2% limit: hemp samples should be declared as non-compliant when THC content, as mean result on a duplicate analysis, exceeds the set limit by 11–15%, depending on THC limit.
Here, we also want to highlight some issues concerning practical and crucial aspects that may arise during hemp analysis.
The sample size recommended by the EU method to be representative of the hemp field is quite large, comprising parts of 50 or 200 plants per field and this may cause difficulty to most analytical laboratories, as samples must be dried within 48 h.
Hemp compliance assessment takes into account the average THC value determined on the representative sample of the field. This means, however, that some single inflorescence, marketed individually, might exceed the legal limit and become a legal question for the owner or the retailer despite it comes from a production assessed as compliant.
By applying the EU method, as any other GC method without derivatization procedures, the evaluation of THCA recovery on own system is advisable, as a possible loss during its thermal conversion into THC may occur.
Currently, only Δ9-THC is the parameter evaluated to allow hemp cultivation. However, recently, new phytocannabinoids were discovered, the Δ9-tetrahydrocannabutol (Δ9-THCB) [56] and the Δ9-tetrahydrocannabiphorol (Δ9-THCP) [57]. The former showed a comparable activity to that of Δ9-THC, while the latter resulted 33 times more active. In the next future, if such high psychotropic effects will be further confirmed, it will be appropriate to include their routine determination.
Finally, it is necessary to carry out inter-laboratory studies on hemp samples with low THC content in order to estimate the actual precision data of the EU method and to suggest, in case, a more suitable official method which will take into account all the analytical issues that may affect the reliability of the results.
References
Small E. Cannabis: a complete guide. Boca Raton: CRC Press; 2017.
Andre CM, Hausman JF, Guerriero G. Cannabis sativa: the plant of the thousand and one molecules. Front Plant Sci. 2016;7:19. https://doi.org/10.3389/fpls.2016.00019.
WHO. Report of the World Health Organization Alcohol, Drugs, and Addictive Behaviours Unit on Cannabis. https://www.who.int/substance_abuse/facts/cannabis/en. Accessed 15 Jan 2021.
Brighenti V, Protti M, Anceschi L, Zanardi C, Mercolini L, Pellati F. Emerging challenges in the extraction, analysis and bioanalysis of cannabidiol and related compounds. J Pharm Biomed Anal. 2020;192:113633. https://doi.org/10.1016/j.jpba.2020.113633.
Citti C, Ciccarella G, Braghiroli D, Parenti C, Vandelli MA, Cannazza G. Medicinal cannabis: principal cannabinoids concentration and their stability evaluated by a high performance liquid chromatography coupled to diode array and quadrupole time of flight mass spectrometry method. J Pharm Biomed Anal. 2016;128:201–9. https://doi.org/10.1016/j.jpba.2016.05.033.
UNO, Single Convention on Narcotics Drugs (1961) https://www.unodc.org/pdf/convention_1961_en.pdf. Accessed 15 Jan 2021.
United Nation Commission on Narcotic Drug. CND votes on recommendations for cannabis and cannabis-related substances, Press Statement – 2 December 2020, https://www.unodc.org/documents/commissions/CND/CND_Sessions/CND_63Reconvened/Press_statement_CND_2_December.pdf. Accessed 15 Jan 2021.
Wikipedia. Legality of cannabis. https://en.wikipedia.org/wiki/Legality_of_cannabis. Accessed 15 Jan 2021.
Small E, Cronquist A. A practical and natural taxonomy for Cannabis. Taxon. 1976;25(4):405–35. https://doi.org/10.2307/1220524.
Government of Western Australia, Department of Justice. Industrial Hemp Act 2004, 22 Sep 2018. https://www.legislation.wa.gov.au/legislation/statutes.nsf/law_a7012_currencies.html. Accessed 15 Jan 2021.
Swiss Confederation. Order of the DFI on the lists of narcotics, of pshycotropic substances, of precursors and chemical adjuvants. OEStup-DFI 30 May 2011. https://www.admin.ch/opc/it/classified-compilation/20101220/index.html. Accessed 15 Jan 2021.
Kruse D. THC regulations regarding industrial hemp in the EU, HempConsult GmbH, 1 August 2016. http://iiha.ie/wp-content/uploads/2018/03/THC-Regulations-regarding-industrial-hemp-in-the-EU-20160901.pdf Accessed 15 Jan 2021.
E.C. Council Directive 2002/53/EC of 13 June 2002 on the common catalogue of varieties of agricultural plant species, Article 17. OJ L 193, 20.7.2002, p. 1–11. http://data.europa.eu/eli/dir/2002/53/oj. Accessed 15 Jan 2021.
E.U. Regulation No 1307/2013 of the European Parliament and of the Council establishing rules for direct payments to farmers under support schemes within the framework of the common agricultural policy and repealing Council Regulation (EC) No 637/2008 and Council Regulation (EC) No 73/2009, Article 32(6). OJ L 347, 20.12.2013, p. 608–670. http://data.europa.eu/eli/reg/2013/1307/oj. Accessed 15 Jan 2021.
E.U. Regulation No 1308/2013 of the European Parliament and of the Council establishing a common organisation of the markets in agricultural products and repealing Council Regulations (EEC) No 922/72, (EEC) No 234/79, (EC) No 1037/2001 and (EC) No 1234/2007, Article 189(1). OJ L 347, 20.12.2013, p. 671–854. http://data.europa.eu/eli/reg/2013/1308/oj. Accessed 15 Jan 2021.
Italian Parliament. Dispositions for the promotion of cultivation and agro-industrial supply chain of cannabis. Law n.242 of 2nd December 2016. https://www.gazzettaufficiale.it/eli/id/2016/12/30/16G00258/sg. Accessed 15 Jan 2021.
President of the Italian Republic. Consolidated text of the laws regarding the regulation of narcotic drugs and psychotropic substances, prevention, treatment and rehabilitation of the relative states of drug addiction. Decree n. 3099 of October 1990. https://www.gazzettaufficiale.it/eli/id/1990/10/31/090G0363/sg. Accessed 15 Jan 2021.
Italian Cannabis producers and retailers. The hemp guide 2020. https://guidacanapa.it/wp-content/uploads/2020/01/magicaitalia2020WEB.pdf. Accessed 15 Jan 2021.
Italian Interior Ministry. Legal-operational aspects related to the phenomenon of commercialization of the inflorescences of textile hemp with low THC content and relations with the legislation on drugs. Note prot.n. 2018/43586 of 20/07/2018.https://www.fuoriluogo.it/wp-content/uploads/2018/09/Ministero-Interno-31-lug-2018-commercializzazione-delle-infiorescenze.pdf. Accessed 15 Jan 2021.
ISO/IEC. Standard n. 17025:2017 General requirements for the competence of testing and calibration laboratories. https://www.iso.org/standards/66912.html. Accessed 15 Jan 2021.
ISO/IEC. Guide 98–3:2008 Uncertainty of measurement - Part 3: Guide to the expression of uncertainty in measurement (GUM:1995). https://www.iso.org/standard/50461.html. Accessed 15 Jan 2021.
ISO/ICS. Standard 21748:2017 Guidance for the use of repeatability, reproducibility and trueness estimates in measurement uncertainty evaluation. https://www.iso.org/standard/71615.html. Accessed 15 Jan 2021.
UNODC. Recommended methods for the identification and the analysis of Cannabis and Cannabis products. United Nations publication, Sales No. E.09.XI.15, ST/NAR/40, 2009, pp. 25–47. https://www.unodc.org/documents/scientific/ST-NAR-40-Ebook_1.pdf. Accessed 15 Jan 2021.
Citti C, Braghiroli D, Vandelli MA, Cannazza G. Pharmaceutical and biomedical analysis of cannabinoids: a critical review. J Pharm Biomed Anal. 2018;147:565–79. https://doi.org/10.1016/j.jpba.2017.06.003.
Cardenia V, Gallina Toschi T, Scappini S, Rubino RC, Rodriguez Estrada MT. Development and validation of a fast gas chromatography/mass spectrometry method for the determination of cannabinoids in Cannabis sativa L. J Food Drug Anal. 2018;26:1283–92.
Aizpurua-Olaizola O, Omar J, Navarro P, Olivares M, Etxebarria N, Usobiaga A. Identification and quantification of cannabinoids in Cannabis sativa L. plants by high-performance liquid chromatography-mass spectrometry. Anal. Bioanal Chem. 2014;406:7549–60.
Gul W, Gul SW, Radwan MM, Wanas AS, Mehmedic Z, Khan II, et al. Determination of 11 cannabinoids in biomass andextracts of different varieties of Cannabis using high-performance liquidchromatography. J AOAC Int. 2015;98:1523–8.
Mandrioli M, Tura M, Scotti S, Toschi TG. Fast detection of 10 cannabinoids by RP-HPLC-UV method in Cannabis sativa L. Molecules. 2019;24:2113. https://doi.org/10.3390/molecules24112113.
Dei Cas M, Casagni E, Saccardo A, Arnoldi S, Young C, Scotti S, et al. The Italian panorama of cannabis light preparation: determination of cannabinoids by LC-UV. Forensic Sci Int. 2020. https://doi.org/10.1016/j.forsciint.2019.110113.
Cannabis Flos. New text of the German pharmacopoeia. Germany: Bonn; 2018.
Citti C, Russo F, Sgrò S, Gallo A, Zanotto A, Forni F, et al. Pitfalls in the analysis of phytocannabinoids in cannabis inflorescence. Anal Bioanal Chem. 2020;412:4009–22. https://doi.org/10.1007/s00216-020-02554-3.
E.C. Union method for the quantitative determination of the Δ9-tetrahydrocannabinol content in hemp varieties. Delegated Regulation (EU) No 639/2014, Annex III as amended by Regulation (EU) 2017/1155. OJ L 167, 30.6.2017, p. 1–15. http://data.europa.eu/eli/reg_del/2017/1155/oj. Accessed 15 Jan 2021.
Italian Customs and Monopolies Agency. Determination of Δ9-tetrahydrocannabinol (Δ9-THC) and other cannabinoids by gas chromatography. MAD 01:2019 rev. 2. https://www.adm.gov.it/portale/-/attivita-dei-laboratori-chimici. Accessed 15 Jan 2021.
Health Canada, Office of Controlled Substances. Industrial Hemp Technical manual.. HECS-OCS-0012004. https://www.canada.ca/content/dam/hc-sc/migration/hc-sc/hc-ps/alt_formats/hecs-sesc/pdf/pubs/precurs/hemp-indus-chanvre/tech-man/hemp-tech-manual-eng.pdf. Accessed 15 Jan 2021.
IUPAC. Harmonised guidelines for single-laboratory validation of methods of analysis. IUPAC technical report. Pure Appl Chem. 2002;74:835–55.
CITAC/EURACHEM. Guide to quality in analytical chemistry. 3rd Edition. 2016. https://www.eurachem.org/index.php/publications/guides/qa. Accessed 15 Jan 2021.
CITAC/ EURACHEM. The fitness for purpose of analytical methods – A laboratory guide to method validation and related topics. Magnusson B, Örnemark U. (eds.) 2nd Edition. 2014. ISBN 978–91–87461-59-0. https://www.eurachem.org/index.php/publications/guides/mv. Accessed 15 Jan 2021.
CITAC/ EURACHEM. Quantifying uncertainty in analytical measurement. Ellison S L. R, Williams A. (eds.), 3rd Edition 2012. ISBN 978–0–948926-30-3. https://www.eurachem.org/index.php/publications/guides/quam. Accessed 15 Jan 2021.
Horwitz W, Albert R. The Horwitz ratio (HorRat): a useful index of method performance with respect to precision. J AOAC Int. 2006;89(4):1095–109.
Iffland K, Carus M, Grotenhermen F. Decarboxylation of Tetrahydrocannabinolic acid (THCA) to active THC. 2016. European Industrial Hemp Association (EIHA). https://eihaorg/media/2014/08/16-10-25-Decarboxylation-of-THCA-to-active-THCpdf Accessed 15 Jan 2021.
Peschel W. Quality control of traditional Cannabis tinctures: pattern, markers, and stability. Sci Pharm. 2016;84(3):567–84. https://doi.org/10.3390/scipharm84030567.
Turner JC, Mahlberg PG. Simple high-performance liquid chromatographic method for separating acidic and neutral cannabinoids in Cannabis sativa L. J Chromatography A. 1982;253:295–303. https://doi.org/10.1016/S0021-9673(01)88392-8.
Dussy FE, Hamberg C, Luginbuhl M, Schwerzmann T, Briellmann TA. Isolation of D9-THCA-A from hemp and analytical aspects concerning the determination of D9-THC in cannabis products. Forensic Sci Int. 2005;149:3–10. https://doi.org/10.1016/j.forsciint.2004.05.015.
Veress T, Szanto JI, Leisztner L. Determination of cannabinoid acids by high-performance liquid chromatography of their neutral derivatives formed by thermal decarboxylation: I. Study of the decarboxylation process in open reactors. J Chromatography A. 1990;520:339–47.
Wang M, Wang YH, Avula B, Radwan MM, Wanas AS, van Antwerp, et al. Decarboxylation study of acidic cannabinoids: a novel approach using ultra-high-performance supercritical fluid chromatography/photodiode array-mass spectrometry, Cannabis. Cannabinoid Res. 2016;1(1):262–71. https://doi.org/10.1089/can.2016.0020.
Repka MA, Munjal M, ElSohly MA, Ross SA. Temperature stability and bioadhesive properties of Δ9-tetrahydrocannabinol incorporated hydroxypropylcellulose polymer matrix systems. Drug Dev Ind Pharm. 2006;32(1):21–32.
Kadis R. Evaluation of the measurement uncertainty: some common mistakes with a focus on the uncertainty from linear calibration. J Chromatogr A. 2017;1499:226–9. https://doi.org/10.1016/j.chroma.2017.03.078.
ISO. Standard n. 5725–2:2019, Accuracy (trueness and precision) of measurement methods and results — Part 2: Basic method for the determination of repeatability and reproducibility of a standard measurement method. https://www.iso.org/standard/69419.html Accessed 15 Jan 2021.
Thompson M, Wood R. The ‘Horwitz ratio’—a study of the ratio between reproducibility and repeatability precisions in the analysis of foodstuffs. Anal Methods. 2015;7:375–9. https://doi.org/10.1039/C4AY02020K.
Royal Society of Chemistry. Is my uncertainty estimate realistic? Analytical Method Committee No. 15 Dec 2003. https://www.rsc.org/images/realistic-estimate-technical-brief-15_tcm18-214874.pdf Accessed 15 Jan 2021.
CITAC/EURACHEM. Use of uncertainty information in compliance assessment. Ellison SLR, Williams A. (Eds.). 1st Edition, 2007. https://www.eurachem.org/index.php/publications/guides/uncertcompliance. Accessed 15 Jan 2021.
ILAC. Guidelines on Decision Rules and Statements of Conformity. G8:09/2019. https://ilac.org/publications-and-resources/ilac-guidance-series/ Accessed 15 Jan 2021.
Joint Committee for Guides in Metrology. Evaluation of measurement data – the role of measurement uncertainty in conformity assessment. JCGM 106: 2012. https://www.bipm.org/utils/common/documents/jcgm/JCGM_106_2012_E.pdf Accessed 15 Jan 2021.
E. C. Report on the relationship between analytical results, measurement uncertainty, recovery factors and the provisions of eu food and feed legislation, with particular reference to community legislation concerning. 2004. https://ec.europa.eu/food/sites/food/files/safety/docs/cs_contaminants_sampling_analysis-report_2004_en.pdf Accessed 15 Jan 2021.
E.C. Commission Decision 2002/657of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results, O J L.22, 17.8.2002, p. 8–36. http://data.europa.eu/eli/dec/2002/657/oj Accessed 15 Jan 2021.
Linciano P, Citti C, Luongo L, Belardo C, Vandelli A, Forni F, et al. Isolation of a high-affinity cannabinoid for the human CB1 receptor from a medicinal Cannabis sativa variety: Δ9-tetrahydrocannabutol, the butyl homologue of Δ9-tetrahydrocannabinol. J Nat Prod. 2020;83(1):88–98.
Citti C, Linciano P, Russo F, Luongo L, Iannotta M, Maione S, et al. A novel phytocannabinoid isolated from Cannabis sativa L. with an in vivo cannabimimetic activity higher than Δ9-tetrahydrocannabinol: Δ9-Tetrahydrocannabiphorol. Sci Rep. 2019;9(1):20335. https://doi.org/10.1038/s41598-019-56785-1.
Acknowledgements
This research work originated in the context of the activities carried out at the II level Master Course in “Chemical and Chemical-Toxicological Forensic Analyses” held at the University of Bologna.
Funding
Open access funding provided by Alma Mater Studiorum - Università di Bologna within the CRUI-CARE Agreement. This work was supported by the “Fundamental and Oriented Research Funding” of the University of Bologna for the years 2019–2020.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Salvatore Sgrò and Benedicta Lavezzi performed the material preparation, data collection, and analysis. The first draft of the manuscript was written by Salvatore Sgrò, the writing–reviewing and editing by Elida Nora Ferri, and all authors commented on the previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The opinions reported in this paper may not entirely reflect and do not imply the Authorities’ position.
Supplementary Information
ESM 1
(PDF 571 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sgrò, S., Lavezzi, B., Caprari, C. et al. Delta9-THC determination by the EU official method: evaluation of measurement uncertainty and compliance assessment of hemp samples. Anal Bioanal Chem 413, 3399–3410 (2021). https://doi.org/10.1007/s00216-021-03283-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-021-03283-x