
Abstract
Aims/hypothesis
The teratogenic consequences of angiotensin-converting enzyme inhibitors angiotensin receptor blockers (ARBs) during the second and third trimesters of pregnancy are well described. However, the consequences of exposure during the first trimester are unclear, especially in diabetes. We report the experience from DIRECT (DIabetic REtinopathy and Candesartan Trials), three placebo-controlled studies designed to examine the effects of an ARB, candesartan, on diabetic retinopathy.
Methods
Over 4 years or longer, 178 normotensive women with type 1 diabetes (86 randomised to candesartan, 32 mg once daily, and 92 assigned to placebo) became pregnant (total of 208 pregnancies).
Results
More than half of patients were exposed to candesartan or placebo prior to or in early pregnancy, but all discontinued it at an estimated 8 weeks from the last menstrual period. Full-term pregnancies (51 vs 50), premature deliveries (21 vs 27), spontaneous miscarriages (12 vs 15), elective terminations (15 vs 14) and other outcomes (1 vs 2) were similar in the candesartan and placebo groups. There were two stillbirths and two ‘sick babies’ in the candesartan group, and one stillbirth, eight ‘sick babies’ and one cardiac malformation in the placebo group.
Conclusions/interpretation
The risk for fetal consequences of ARBs in type 1 diabetes may not be high if exposure is clearly limited to the first trimester. Long-term studies in fertile women can be conducted with ARBs during pregnancy, provided investigators diligently stop their administration upon planning or detection of pregnancy.
Trial registration
ClinicalTrials.gov DIRECT-Prevent 1 NCT00252733; DIRECT-Protect 1 NCT00252720; DIRECT-Protect 2 NCT00252694.
Funding
The study was funded jointly by AstraZeneca and Takeda.
Similar content being viewed by others


Introduction
Agents that block the renin–angiotensin system (RAS), angiotensin-converting enzyme inhibitors (ACEis) and blockers of the angiotensin type-1 (AT1) receptor (ARBs), are among the most utilised and effective antihypertensive agents. In particular, RAS blockers are important for patients with type 1 and 2 diabetes with hypertension and/or microalbuminuria because they reduce progression of diabetic renal damage [1]. However, both ACEis and ARBs are known to cause fetal malformations and neonatal problems if administered during pregnancy and this can prove a deterrent to their use in women of child-bearing potential [2, 3].
The teratogenic potential of RAS-inhibiting/blocking agents administered during the second and third trimesters of pregnancy is well established. Abnormalities reported include fetal craniofacial abnormalities and limb contractures, probably consequent to oligohydramnious and failed renal development [3]. However, whether there are similar deleterious effects of exposure to ACEis or ARBs during the first trimester remains controversial, with conflicting reports of increased malformations, specifically cardiac malformations, in infants of mothers exposed to ACEis, as opposed to other antihypertensive agents, during the first 3 months of pregnancy [4–6]. The controversy is further complicated because both hypertension [6, 7] and diabetes [8] carry increased teratogenic potential per se, which makes it difficult to discern the effects of either disease from those of the drugs. In addition, previous studies [4, 5] made a specific effort to exclude women who had diabetes before pregnancy from their analyses. The absence of data specific to diabetic women of child-bearing age presents a difficult dilemma. Should young diabetic women with hypertension or early diabetic nephropathy be denied treatment with RAS-inhibiting agents for fear of fetal exposure in case of pregnancy? In addition, it is important to include rather than exclude women of child-bearing potential from clinical trials of these agents, so that the results can be generalised to young diabetic patients of both sexes.
DIRECT (DIabetic REtinopathy and Candesartan Trials) [9, 10], a group of clinical studies designed to study the effects of candesartan on diabetic retinopathy, offers a unique opportunity to investigate the consequences of ARB exposure during early pregnancy because: (1) women with diabetes were randomised to treatment with either candesartan or placebo for at least 4 years; (2) the study protocol included careful instructions to stop the test drug whenever pregnancy was either being contemplated or otherwise diagnosed; and, (3) all women who became pregnant were normotensive. As none of the women who became pregnant took the study drug beyond the eighth week, the reports collected are likely to reflect exposure during the early first trimester only.
Methods
The design of the DIRECT programme has been reported elsewhere [9, 10]. In brief, 1,421 patients with type 1 diabetes and no retinopathy at baseline (DIRECT-Prevent 1), 1,905 patients with type 1 diabetes and mild to moderately severe non-proliferative retinopathy at baseline (DIRECT-Protect 1) and 1,905 patients with type 2 diabetes and mild to moderately severe non-proliferative retinopathy (DIRECT-Protect 2) were randomised to either candesartan, 32 mg once daily, or placebo. All patients were normoalbuminuric (albumin excretion rate <20 μg/min) at baseline. Type 1 patients were all normotensive (BP ≤ 130/85 mmHg) and type 2 patients were either normotensive, or mildly hypertensive (BP ≤ 160/90 mmHg) and treated with agents other than RAS inhibitors. All subjects gave their informed consent. The study protocol was approved by the responsible ethics committee in each participating centre and carried out in accordance with the Declaration of Helsinki as revised in 2000.
Women of child-bearing potential were advised to practice effective contraception. Investigators were instructed to halt study drug on first knowledge of pregnancy or if a woman was planning pregnancy, to report the instance with use of a specific pregnancy report form, and later to report the outcome of pregnancy on a pregnancy outcome form. Where required by local regulations and/or the protocol, the investigators also reported pregnancy outcome-related adverse events. The pregnancy report form included the dates of the last menstrual period, expected date of delivery and last dose of study drug. Pregnancy outcome forms included data on delivery (date, pre-term or full, medical or spontaneous termination) and child status (healthy, sick, congenital malformation, stillborn). Premature birth was defined as birth less than 37 weeks from the last menstrual period. A description of diseases in the infants was not requested by the form. The DIRECT database included data on baseline clinical variables, such as BMI, BP, HbA1c and creatinine. Finally, the dates of re-entry into the study after end of pregnancy and/or lactation were recorded. The data from the pregnancy report and pregnancy outcome forms of women who received at least one dose of the study drug were compiled and aligned with data from the studies, including adverse event reports. Alignment between data was established through use of calendar dates, as the pregnancies occurred without relation to scheduled protocol assessments.
Estimates of the interval between study entry and beginning of pregnancy were based on the assessed last menstrual period.
Results
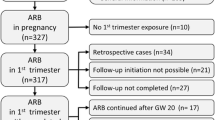
Overall, 615 (43.3%), 813 (42.7%) and 957 (50.2%) women were randomised to DIRECT-Prevent 1, DIRECT-Protect 1 and DIRECT-Protect 2, respectively. Of the women who were randomised and took at least one dose of study drug, 178 patients (73 from Prevent 1 and 105 from Protect 1) became pregnant (86 from the candesartan and 92 from the placebo groups) (Fig. 1). These 178 patients had a total of 208 pregnancies (12 patients in the candesartan and 12 in the placebo group had more than one pregnancy). No pregnancies occurred in DIRECT-Protect 2 (mean age 57.9 years). The baseline characteristics of women in DIRECT-Prevent 1 and DIRECT-Protect 1 are shown in Table 1. The majority of patients were white.

Distribution of pregnancies in DIRECT–Prevent 1, DIRECT–Protect 1 and DIRECT–Protect 2 (safety population, at least one dose of study drug taken)
Compared with those who did not become pregnant, women who did were younger and tended to have shorter durations of diabetes, but were otherwise similar. Patient characteristics were also similar in the candesartan and placebo groups.
Considering the first pregnancy during the study in each woman who became pregnant, 14 women taking candesartan and 16 taking placebo discontinued the study drug well in advance of the last menstrual period (more than 8 weeks before); 18 women taking candesartan and 13 taking placebo discontinued the drug 8 to 0 weeks prior to last menstrual period; 42 women taking candesartan and 45 taking placebo discontinued 0 to 8 weeks after their last menstrual period. In 30 women the interval between the last dose and last menstrual period could not be numerically calculated, but all patients were known to have discontinued candesartan treatment before the end of the first trimester. Accordingly, over half the patients were likely to have been exposed to study drug during trimester 1. Evaluation of all subsequent pregnancies showed that candesartan treatment was either stopped before the end of the first trimester or was never resumed after the preceding pregnancy.
Delivery outcomes were similar for the candesartan and placebo groups: full-term delivery (51 candesartan, 50 placebo), premature birth (21 candesartan, 27 placebo), spontaneous miscarriage (12 candesartan, 15 placebo), elective termination (15 candesartan, 14 placebo), other (one candesartan, two placebo). Most infants were healthy, whether full term or premature. There were two stillbirths in the candesartan group and one in the placebo group, two ‘sick babies’ in the candesartan group and eight in the placebo group. The only congenital malformation reported was a ventricular septal defect in the placebo group.
No geographic pattern appeared for distribution of pregnancies, with different frequencies of pregnancies across countries being accounted for by the numbers of patients included in DIRECT-Prevent 1 and DIRECT-Protect 1 in the different countries. A total of 69 patients restarted the study drug and the median time between last menstrual period and resumption of study drug was 406 days for the candesartan and 456 days for the placebo patients. The time to re-entry into the studies varied considerably, depending on any period of lactation in addition to pregnancy, because candesartan is contraindicated during lactation. Pregnancies not going full term were associated with the women rapidly resuming study drug.
Discussion
The DIRECT experience indicates that exposure to a relatively high dosage of candesartan, 32 mg/day, for up to 8 weeks into the first trimester of pregnancy may not result in a higher rate of malformations than placebo in normotensive normoalbuminuric women with type 1 diabetes. Only one instance of cardiac malformation was reported in the study and it occurred in a woman administered placebo, taking no antihypertensive drugs and using only insulin as a glucose-lowering agent.
While the fetal consequences of RAS-inhibiting drugs seem to result primarily from exposure during the second and third trimesters, to date there have been insufficient data to exclude similar detrimental effects of exposure during the first trimester. In a cohort study of 29,507 infants born in Tennessee, USA, Cooper et al. [4] reported that 202 infants exposed to an ACEi during the first trimester had an increased risk of malformations, specifically cardiac malformations, compared with 209 infants whose mothers took other antihypertensive agents during the first trimester. In a Swedish cohort of 1,418 women who had taken antihypertensives during the first trimester, Lennestål et al. [5] were unable to find a different incidence of cardiac malformations between the infants of mothers who had taken an ACEi and those who had used other agents, notably beta-blockers. Caton et al. reported similar rates of use of any antihypertensive medication, including RAS blockers, in the mothers of 5,021 infants born with cardiac malformations and 4,796 controls without birth defects in a survey across ten US states covering the years 1997–2003 [6].
The DIRECT experience is pertinent in this regard. Although small compared with the above observational studies, the clinical trial setting allowed for a fairly precise estimation of drug exposure relative to conception date and for comparison with placebo. It is estimated that the frequency of fetopathy from drug-related RAS inhibition consequent to exposure during the second and third trimesters is about 10–20% [11]. If the risk were of similar magnitude in the first trimester, one would have expected several cases to occur in the candesartan-treated group.
Estimates of the interval between study drug discontinuation and beginning of pregnancy were based on the assessed last menstrual period. Cross-matching of the pregnancy report form and DIRECT database indicates that no woman took the study drug after the eighth week of pregnancy. Some uncertainty as to the precise date of conception remains inevitable, even though the last menstrual period is the accepted method for establishing it. However, these data strongly suggest that there was exposure to study drug during the first trimester in many of the women taking candesartan, but in no case did such exposure extend beyond the first trimester.
Overall, the incidence of cardiac malformations in DIRECT was lower than those reported in previous studies of hypertensive non-diabetic women [2–6]. This suggests that hypertension per se might have played a teratogenic role in those case series independently of the agents used to treat it. Even taking into account the potential teratogenic effect of diabetes and the fact that HbA1c at enrolment and prior to last menstrual period exceeded recommended targets [12], the number of adverse pregnancy outcomes was fairly low in the DIRECT experience.
It is important to note, however, that both receptor types AT1 and AT2 are produced early in the human embryo (24 days’ gestation) and all components of the RAS have been demonstrated by 30–35 days of gestation [13], suggesting a role for angiotensin II in early organogenesis. In spite of explicit warning, women of fertile age continue to be exposed to RAS-inhibiting/blocking agents throughout pregnancy with unfortunate consequences [4–6, 14]. The treatment of hypertension and diabetic nephropathy in non-pregnant women with type 1 diabetes mellitus is clearly justified, at least until pregnancy is sought or confirmed. However, in real life women may present much later than the eighth week, even when planning pregnancy, and may not seek medical advice early. Hence, the need to be extremely careful when using RAS blockers in fertile women.
The DIRECT experience supports the notion that benefits vs risks of treating diabetic women of child-bearing potential can be verified by including these patients into clinical trials that assess the effects of RAS blockers. In DIRECT there was little concern that the interruption in study drug necessitated by pregnancy would influence the overall study results as this situation applied to only a fraction of the study population and most patients resumed study drug at the conclusion of pregnancy/lactation.
In conclusion, the DIRECT clinical trial experience indicates that: (1) the teratogenic risks of ARBs may be small if exposure is clearly limited to early first trimester of pregnancy; and (2) women of child-bearing age can take RAS-inhibiting/blocking agents for valid reasons and still avoid exposure during the vulnerable (teratogenic risk) periods of pregnancy if both patients and prescribing physicians are appropriately advised and provided they will diligently stop the agent upon planning or detection of pregnancy. However, longer surveys of fetal exposure to RAS blockers are required to confirm that the risk is as low as reported in DIRECT, and other trials in which pregnancies occurred should report their outcomes in order that a subsequent meta-analysis may be performed. Appropriately dimensioned trials of antihypertensive and other therapies during pregnancy might provide important information and guidance for women with diabetes.
Abbreviations
- ACEi:
-
Angiotensin-converting enzyme inhibitor
- ARB:
-
Angiotensin type-1 receptor blocker
- AT1/2 :
-
Angiotensin type-1/-2
- DIRECT:
-
Diabetic Retinopathy and Candesartan Trials
- RAS:
-
Renin–angiotensin system
References
Schjoedt KJ, Hansen HP, Tarnow L, Rossing P, Parving HH (2008) Long-term prevention of diabetic nephropathy: an audit. Diabetologia 51:956–961
Alwan S, Polifka JE, Friedman JM (2005) Angiotensin II receptor antagonist treatment during pregnancy. Birth Defects Research (Part A) 73:123–130
Quan A (2006) Fetopathy associated with exposure to angiotensin converting enzyme inhibitors and angiotensin receptor antagonists. Early Human Development 82:23–28
Cooper WO, Hernandez-Diaz S, Arbogast PG et al (2006) Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med 354:2443–2451
Lennestål R, Otterblad Olausson P, Källén B (2009) Maternal use of antihypertensive drugs in early pregnancy and delivery outcome, notably the presence of congenital heart defects in the infants. Eur J Clin Pharmacol 65:615–625
Caton AR, Bell EM, Druschel CM et al (2009) Antihypertensive medication use during pregnancy and the risk of cardiovascular malformations. Hypertension 54:63–70
Yoder SR, Thornburg LL, Bisognano JD (2009) Hypertension in pregnancy and women of childbearing age. Am J Med 122:890–895
Allen VM, Armson BA, Wilson RD et al (2007) Teratogenicity associated with pre-existing and gestational diabetes. J Obstet Gynaecol Can 29:927–944
Chaturvedi N, Porta M, Klein R et al (2008) Effect of candesartan on prevention (DIRECT–Prevent 1) and progression (DIRECT–Protect 1) of retinopathy in type 1 diabetes: randomised, placebo-controlled trials. Lancet 372:394–402
Sjølie AK, Klein R, Porta M et al (2008) Effect of candesartan on progression and regression of retinopathy in type 2 diabetes (DIRECT–Protect 2): a randomised placebo-controlled trial. Lancet 372:1385–1393
Food and Drug Administration (1992) Dangers of ACE inhibition during second and third trimesters of pregnancy. Med Bull 22:2
American Diabetes Association (2010) Standards of medical care in diabetes—2010. Diabetes Care 33(Suppl 1):S11–S61
Schultz S, Moullec J-M, Corvol P, Gasic J-M (1996) Early expression of all the components of the renin–angiotensin system in human development. Am J Pathology 149:2067–2079
Haaland K (2010) Angiotensin II receptor antagonists against migraine in pregnacny: fatal outcome. J Headache Pain 11:167–169
Duality of interest
M. Porta, R. Bilous, N. Chaturvedi, J. H. Fuller, R. Klein, T. Orchard, H.-H. Parving, A.-K. Sjølie have received consultancy fees for attending committee meetings. M. Porta, A.-K. Sjølie and R. Bilous received fees as national coordinators, and M. Porta, N. Chaturvedi, R. Bilous, A.-K. Sjølie, J. H. Fuller and H.-H. Parving have received honoraria for scientific presentations from AstraZeneca and Takeda. J. W. Hainer, S.-O. Jansson and A. Malm are employees of AstraZeneca.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Members of the Study Group are shown in the Electronic supplementary material (ESM).
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
PDF 68 kb
Rights and permissions
About this article
Cite this article
Porta, M., Hainer, J.W., Jansson, SO. et al. Exposure to candesartan during the first trimester of pregnancy in type 1 diabetes: experience from the placebo-controlled diabetic retinopathy candesartan trials. Diabetologia 54, 1298–1303 (2011). https://doi.org/10.1007/s00125-010-2040-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-2040-1