
Abstract
Aims/hypothesis
A local angiotensin-generating system has been found in the exocrine pancreas. This study aimed, primarily, to investigate the existence of a local angiotensin-generating system in the pancreatic islets and, secondly, to elucidate its role in regulating insulin secretion.
Methods
Real-time RT-PCR and western blot were used to investigate if angiotensin-generating components are present in the mouse pancreatic islets, which are subject to regulation by islet transplantation. The localisation of AT1-receptors in islets was investigated by immunohistochemistry. Batch-type incubations of isolated islets were applied for studying the influence of angiotensin II on the glucose-stimulated insulin release, glucose oxidation and (pro)insulin, and total protein biosynthesis.
Results
Major components, namely angiotensinogen, ACE, AT 1 - and AT 2 -receptors, were expressed in endogenous islets. AT1-receptors were localised to pancreatic beta cells. Exposure of the isolated islets to angiotensin II induced a dose-dependent inhibition of glucose-stimulated insulin release and inhibited (pro)insulin biosynthesis. This inhibitory action was fully preventable by pretreatment of the islets with losartan, an AT1-receptor antagonist. We also investigated if the expression of these components was changed after islet transplantation. Notably, a markedly increased expression of mRNA for the AT 1 -receptor was observed in islets retrieved from 4-week-old syngeneic islet transplants, a finding that was confirmed at the protein level.
Conclusion/interpretation
These data indicate the existence of an islet angiotensin-generating system of potential importance in the physiological regulation of glucose-induced insulin secretion, thus diabetes mellitus. The increased expression of the AT1-receptor in islet transplants could have relevance to islet-graft function.
Similar content being viewed by others

The renin-angiotensin system (RAS) has long been known best for its haemodynamic regulation by means of two major angiotensin II (Ang II) receptors, AT1 and AT2 [1]. The two definitive enzymes for this system are renin and angiotensin-converting enzyme (ACE), which determine the generation of the physiologically active peptide, Ang II. The biosynthetic pathways for this classic cascade to produce Ang II and several bioactive peptides such as Ang III, Ang IV and Ang (1–7) are schematically illustrated in Fig. 1. However, alternate enzymes to renin and ACE including kallikrein, tonin and chymase can generate Ang II directly from its precursor angiotensinogen (Ao) rather than the definitive enzymatic cascade of Ang II generation [2]. It is therefore more appropriate to describe the RAS as an “angiotensin-generating system”, which consists of obligatory components (Ao, Ang II and its receptors), linked by non-exclusive components (renin and ACE).

Outline of the biosynthetic pathways of the renin-angiotensin system. In this system, the precursor angiotensinogen is an obligatory component while renin and ACE are two important enzymes for the cascade. ACE, angiotensin-converting enzyme; ACE-2, a homologue of ACE; APB, aminopeptidase B; APN, aminopeptidase N; NEP, neutral endopeptidase; PEP, propylendopeptidase
In recent years, the existence of a local angiotensin-generating system in multiple tissue organs has been shown [3, 4]. This implies that locally produced Ang II exerts local actions from such diverse targets as the heart [5], adrenals [6] and gonads [7] to the pancreas, recently reviewed [8]. Previous studies have shown the presence of such a local system in the pancreas of various species, including the dog [9], rat [10, 11], mouse [12] and human [13]. Available data suggest some potential roles for the angiotensin-generating system in the exocrine and endocrine pancreas [14], such as regulation of ductal anion secretion in the exocrine pancreas [15]. Nevertheless, the potential existence and role of an angiotensin-generating system in the pancreatic islets remain largely unexplored. Indications for a role of Ang II in islets were provided by a perfusion study of whole pancreata, whereby the first phase of glucose-stimulated insulin release was markedly impaired by Ang II [16]. However, this finding was solely attributable to the potent vasoconstrictor action of Ang II, which caused a decrease of islet blood flow in the pancreas.
The present study, therefore, aimed specifically to investigate the existence of an angiotensin-generating system in the pancreatic islets; if so, to elucidate its role in the regulation of insulin secretion and its biosynthesis by studies of isolated pancreatic islets. On the other hand, components of the angiotensin-generating system in the pancreas have previously been shown to be up-regulated by chronic hypoxia [17, 18]. In view of the sustained decrease of oxygen tension in pancreatic islet transplants [19, 20, 21], we also investigated the expression of angiotensin-generating components in retrieved transplanted islets.
Materials and methods
Animals
Male C57BL/6J mice were used in all experiments. The animals were provided by the Laboratory Animal Services Centre of the Chinese University of Hong Kong. The experimental procedures were approved by the Animal Experimentation Ethics Committee of the Chinese University of Hong Kong and followed the “Principles of laboratory animal care” (NIH publication no. 85-23, revised 1985).
Pancreatic islet isolation
Islets were isolated from the pancreas as described previously [22]. Briefly, C57BL/6J mice aged 10 weeks were killed by cervical dislocation and the pancreata were dissected out. The pancreata were then placed in cold Hanks’ solution (Sigma Aldrich, St. Louis, USA) and injected with Hanks’ solution by means of a 27 G syringe. The inflated pancreata were cut into small pieces with similar size. These pieces were transferred to vials containing collagenase solution (25 mg/8 ml Hanks) (Roche Molecular Biochemicals, Mannheim, Germany). The vials were shaken vigorously at 37°C for approximately 15 min. The collagenase digestion was terminated by the addition of cold Hanks’ solution and the digest was then washed three times by filling the vial with Hanks. The islets were then picked using mouth-pipettes and cultured free-floating for 4 to 7 days in non-adherent culture dishes using RPMI 1640 medium (Sigma Aldrich, St Louis, USA) supplemented with 10% (vol/vol) fetal bovine serum (Sigma Aldrich). Medium was changed every other day.
Measurements of insulin release
Groups of ten islets were transferred in duplicate to Falcon 24-well culture plates containing 0.25 ml Krebs-Ringer bicarbonate buffer (KRBB) supplemented with 10 mmol/l HEPES and 2 mg/ml of BSA. For the first hour of incubation at 37°C (O2/CO2, 95:5), the medium contained 1.7 mmol/l glucose. The medium was then removed and replaced with 0.25 ml of KRBB containing 16.7 mmol/l glucose and incubated for another hour [23]. The effect of Ang II (Sigma Aldrich) on insulin release from isolated islets was determined at 0.1, 1, 10 and 100 nmol/l of Ang II. Ang II was added in the KRBB medium with 16.7 mmol/l glucose during the second hour of incubation. Specific Ang II-receptor antagonists for AT1, losartan (Merck & Co., N.J., USA; 1 µmol/l) and for AT2, PD123319 (Sigma, St. Louis, Mo., USA; 1 µmol/l) were administered 10 min before the Ang II treatment. After the incubation, the medium was collected for the measurement of insulin release using a mouse insulin ELISA kit (Mercodia, Uppsala, Sweden).
Measurements of islet glucose oxidation rate
Triplicate groups of ten islets were transferred to glass vials containing 100 µl of KRBB supplemented with D-[U-14C]glucose (Amersham-Pharmacia Biotech, Uppsala, Sweden) and non-radioactive glucose to a final glucose concentration of 1.7 or 16.7 mmol/l glucose, respectively (spec. act. 0.5 mCi/mmol). To some vials Ang II was added to a final concentration of 100 nmol/l. The islet glucose oxidation rates during 90-min incubation at 37°C (95%O2/5%CO2) were measured as described elsewhere [24].
Measurements of (pro)insulin and total protein biosynthesis
Duplicate groups of ten islets were incubated, with or without 100 nmol/l Ang II, at 37°C in 100 µl of KRBB containing 50 µCi/ml of L-[4,5–3H]-leucine (Amersham-Pharmacia) and 1.7 or 16.7 mmol/l glucose in an atmosphere of humidified air plus 5% CO2. After 2 h, the islets were washed in buffer containing non-radioactive leucine (10 mmol/l) and sonicated in 200 µl of redistilled water. The amount of labelled (pro)insulin was measured by an immunoabsorption technique [25], and the total protein biosynthesis was measured in trichloroacetic acid precipitates of the islet homogenate.
Pancreatic islet transplantation
The method for islet transplantation has been described previously [19, 20, 21, 26]. Briefly, three hundred isolated islets were packed into the tip of the mouth-pipette after 4 to 7 days of culture. A recipient C57BL/6J mouse was anaesthetised with an i.p. injection of 2.5% (wt/vol) pentobarbital and a small incision was made on the left renal capsule. The tip of the mouth-pipette was introduced between the renal parenchyma and the capsule followed by gentle ejection of the pellet. Four weeks after transplantation, the mice were killed and the islet transplant was dissected out. As control, pancreatic islets isolated from animals matched for age were used.
Real-time RT-PCR analysis
Real-time quantitative RT-PCR was carried out using an ABI PRISM 7700 Sequence Detection System (PE Applied Biosystems, Foster City, Calif., USA). Total RNA was extracted from pools of all islets isolated from control and retrieved from transplanted mice (groups of 8–10 mice each), using the TRIzol reagent kit (Life Technologies, Gaithersburg, Md., USA) according to the manufacturer’s instructions. Total RNA was also extracted from liver and kidney to study the relative abundance of islet mRNA compared to these tissues. The RNA was used as template in one-step Taq Man amplification reactions. TaqMan primers and probes for Ao, ACE, AT 1 and AT 2 were designed from mouse cDNA sequence using Primer Express Software purchased from Applied Biosystems Perkin-Elmer. Mouse β-actin RNA was used as internal control. TaqMan reactions were set up in a reaction volume of 25 µl by using components supplied in a Taq Man PCR reagent kit. Each reaction consisted of 12.5 µl PCR master mix, 18 µmole of each amplification primer, 25 µmol/l corresponding Taq Man probe and 30 ng RNA template. Each sample was run in duplicate with initial 30 min at 48°C and 10 min at 95°C for reverse transcription, followed by 40 cycles at 95°C for 15 s and at 60°C for 1 min. Amplification data were collected by the 7700 Sequence Detector and analysed by using the Sequence Detection System software. The RNA concentration was determined from the threshold cycle (CT) at which fluorescence is first detected, the cycle number being inversely related to RNA concentration. The brief details of RAS sequence of PCR primers and their specific probes, as well as corresponding cDNA sequence, were listed in Table 1. The fold changes in mRNA for RAS by islet transplantation were calculated using the 2−ΔΔCT method, as described in the section of statistical data analysis.
Western blot analysis
Islets were isolated from control or retrieved from transplanted mice. Protein of 300 islets was extracted using CytoBuster Protein Extraction Reagent (Novagen, Darmstadt, Germany). Protein content of lysates was determined by a Bio-Rad protein assay kit (Bio-Rad, Munich, Germany). Western blot analysis of AT1-receptor protein expression was done according to our previously described protocol [27]. Briefly, proteins (10 µg/lane) were subject to electrophoresis on a 12% (wt/vol) polyacrylamide. The blotted protein was saturated with 5% (wt/vol) of skimmed milk in phospate-buffered saline (PBS; pH 7.4) and 0.1% (vol/vol) of Tween 20 for 1 h at room temperature. The membrane was sequentially incubated in rabbit anti-AT1 serum (1:300 dilution) overnight at 4°C and a peroxidase-labelled anti-rabbit IgG antibody (1:1300 dilution) for 1 h at room temperature. The positive signal was revealed using ECL plus western blotting detection reagents and autoradiography film (Amersham, Buckinghamshire, UK). The chemiluminescence intensity of the bands was quantified using an image analyser (Molecular Dynamics Image Quant, Sunnyvale, Calif., USA).
Immunohistochemical localization of AT1 receptor
Immunohistochemistry coupled with a double-staining technique [28, 29] was used to determine the specific localisation of AT1-receptor in islets. Isolated islets were fixed with 4% (vol/vol) cold paraformaldehyde and embedded in paraffin. Sections (5 µm thick) were mounted on glass slides, deparaffinized and processed for indirect immunofluorescent double staining. After several washes with PBS (pH 7.4), each slide was incubated with 4% (wt/vol) Normal Donkey serum (Jackson ImmunoResearch, Pa., USA) for 60 min at 37°C. Excess blocking solution was removed and the slide was incubated overnight at 4°C with rabbit anti-AT1 serum (Santa Cruz Biotech., Santa Cruz, Calif., USA), diluted to 1:25, and with goat anti-insulin serum (Santa Cruz Biotech.), diluted to 1:50. After three washes with PBS, the primary antibodies were detected using an anti-rabbit antibody labelled with Rhodamine (AT1-receptor) and an anti-goat antibody labelled with Aminomethylcoumarin Acetate (insulin) (Jackson ImmunoResearch) at room temperature for 1 h. Preadsorption and omission of primary antibodies were used as negative controls. Positive immunostaining for AT1 (red) and for insulin (blue) was examined with a fluorescent microscope equipped with a DC 200 digital camera (Leica Microsystems).
Statistical data analysis
Results were expressed as means ± SEM for all groups. Multiple comparisons between data were done using ANOVA followed by Tukey’s test, or, when comparisons only were made toward control, by Dunnett’s test. When only two groups were compared, probabilities (p) of chance differences between the experimental groups were calculated with Student’s unpaired two-tailed t test. For all comparisons, p<0.05 was considered statistically significant. For Western blot analysis, data were normalised as a percentage of control. For real-time RT-PCR, the relative expression was normalised as percentage of β-actin and calculated using the comparative CT method of 2−ΔΔCT, as described previously [30, 31].
Results
Ang II and insulin release from isolated islets
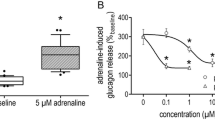
Insulin release from the isolated islets was markedly enhanced as expected when changing glucose concentration in the incubation medium from 1.7 to 16.7 mmol/l (Fig. 2a). However, the glucose-stimulated insulin release was dose-dependently decreased by adding 0.1, 1, 10 and 100 nmol/l of Ang II (Fig. 2a). At the highest concentration of Ang II used (100 nmol/l), the glucose-induced insulin release could be completely prevented. Pretreatment of isolated islets with 1 µmol/l of losartan, a specific antagonist for the AT1-receptor, before adding Ang II (100 nmol/l) completely restored the insulin secretion to the glucose-stimulated level (Fig. 2b). However, pretreatment with 1 µmol/l of PD123319, a specific antagonist for the AT2-receptor had no effect on glucose-stimulated insulin release. In contrast, losartan per se had no effect on glucose-stimulated insulin secretion.

(a) Insulin release from isolated mouse islets in the presence of 1.7 (low; L) or 16.7 mmol/l (high; H) glucose. Ang II was applied at concentrations of 0.1, 1, 10 and 100 nmol/l at the higher glucose concentration. (b) Effects of losartan (Los, 1 µmol/l) and Ang II (100 nmol/l) on the glucose (16.7 mmol/l)-stimulated insulin release from isolated islets. All data are expressed as means ± SEM for four experiments in each group. *denotes p<0.05 when compared to islets exposed to 16.7 mmol/l glucose only
Ang II and islet glucose oxidation rate, (pro)insulin and total protein biosynthesis
Islet glucose oxidation rate, (pro)insulin and total protein biosynthesis were, as the insulin release, markedly higher at 16.7 mmol/l than 1.7 mmol/l glucose (Fig. 3 and Fig. 4). Addition of Ang II (100 nmol/l) to the islets during incubation did not affect either the islet glucose oxidation rate at 1.7 or 16.7 mmol/l glucose (Fig. 3). Islet (pro)insulin biosynthesis at 16.7 mmol/l glucose but not at 1.7 mmol/l glucose was, however, inhibited by 100 nmol/l Ang II (Fig. 4a). Islet total protein synthesis was not affected by Ang II either at 1.7 or 16.7 mmol/l glucose (Fig. 4b).

Influence of Ang II (100 nmol/l) on the glucose oxidation rate in isolated mouse islets exposed to 1.7 (low; L) or 16.7 mmol/l (high; H) glucose. All data are expressed as means ± SEM for nine experiments in each group

Influence of Ang II (100 nmol/l) on the (pro)insulin (a) and total protein (b) biosynthesis in isolated mouse islets exposed to 1.7 or 16.7 mmol/l glucose. All data are expressed as means ± SEM for six experiments in each group. * denotes p<0.05 when compared to corresponding islets exposed to 1.7 mmol/l glucose, whereas # denotes p<0.05 when compared to islets exposed to 16.7 mmol/l glucose only
Expression of RAS components in endogenous and transplanted islets
The expression of several major components, namely AT 1 - and AT 2 -receptors, ACE and Ao, were examined and compared using real-time quantitative RT-PCR coupled with the 2−ΔΔCT comparative method in endogenous and transplanted islets. mRNAs for all the components studied were expressed in endogenous islets (Fig. 5). When compared to endogenous islets, the relative expression of mRNAs for AT 1 to β-actin was increased by about threefold in transplanted islets (Fig. 5a). In contrast, the relative expression of mRNAs for AT 2 to β-actin was down-regulated to approximately one third in transplanted islets (Fig. 5b). The expression of mRNA for ACE in endogenous islets did not differ significantly from transplanted islets (Fig. 5c) whereas the expression of angiotensinogen mRNA in transplanted islets was lower than in endogenous islets (Fig. 5d). As calculated from the CT (Table 2), the relative abundance of different mRNA studied was 0.9, 1.1 and 1.2 for AT2, Ao and ACE, respectively as compared to AT 1 expression being as 1. On the other hand, the relative abundance of AT 1 and AT 2 expression in the mouse islets was about 70%, when compared with that in the liver and kidney based on the calculation from the CT using β-actin as internal control.

Real-time RT-PCR analysis of the mRNA expression of (a) AT 1 receptor; (b) AT 2 receptor; (c) ACE and (d) angiotensinogen in endogenous and transplanted mouse islets. The relative expression was normalised as percentage of β-actin calculated using the comparative CT method of 2−ΔΔCT (Table 2). All data are expressed as means ± SEM for four experiments in each group. * denotes p<0.05 when compared to endogenous islets
Expression of AT1-receptor protein in endogenous and transplanted islets
Western blot analysis was carried out to confirm that findings of AT1-receptor mRNA in endogenous and transplanted islets were reflected also at the protein level (Fig. 6). A major protein band of about 50 Mr was expressed both in endogenous and transplanted islets; in the latter case this was a band of strong intensity (Fig. 6a). This molecular mass (50 Mr) was in close agreement with that of AT1-receptor protein in mouse kidney, which was used as a positive control for the expression of the AT1-receptor. The relative expression of AT1-receptor protein in transplanted islets was up-regulated twofold when compared to that in endogenous islets, as determined by image analysis (Fig. 6b).

Western blot analysis of AT1 receptor protein from endogenous and transplanted mouse pancreatic islets. (a) Representative gel image. Lane 1 shows protein of the AT1 receptor from mouse kidney. A major band of about 50 Mr was detected. Lanes 2 and 3 show the expression of the AT1 receptor in protein of 10 µg from isolated endogenous and transplanted islets, respectively. (b) The relative expression of the AT1 receptor protein in transplanted islets when compared to endogenous islets. All data are expressed as means ± SEM for four experiments. * denotes p<0.05 when compared to endogenous islets
Localization of AT1 receptors in isolated pancreatic islets
A technique with double immunostaining was used for precise localisation of AT1-receptors in the pancreatic islets (Fig. 7). The localisation of AT1-receptors (Fig. 7a) to the insulin secreting beta cells (Fig. 7b) was identified by an overlay of immunoreactivity for insulin and the AT1-receptor (Fig. 7c). Specificity of the immunoreactivity was validated by control experiments either in preadsorption of AT1 receptor with its blocking peptide in excess (Fig. 7d) or in omission of primary antibody for the AT1-receptor.

Immunohistochemical localisation of AT1 receptors and insulin in isolated mouse pancreatic islets. (a) Isolated islet stained for AT1 receptors (red). (b) Islet stained for insulin (blue). (c) Immunoreactivity for that AT1 receptor was co-localised with that for insulin in the islet beta-cells (purple). (d) Negative control with the preadsorption of the primary antibodies with its excess blocking peptide. Magnification: 40×. Bar=40 µm
Discussion
Our study provides evidence for the existence of an angiotensin-generating system in the islets by showing the expression of several components (AT 1 and AT 2 receptors, ACE, and Ao) in the pancreatic islets. Of particular importance in this context is the presence of Ao, which constitutes a mandatory component for a local angiotensin-generating system. The existence of such a system makes a local production of Ang II probable, which produces its concentrations several times higher than those measured in peripheral blood. Indeed, islet blood flow seems to be suppressed by such locally produced Ang II during normal conditions [8, 16]. More importantly, Ang II induced a marked and dose-dependent decrease of glucose-stimulated insulin release from isolated islets in the present study, indicating that Ang II plays an important role in regulating islet insulin secretion. With the highest dose of Ang II used (100 nmol/l), the glucose-stimulated insulin secretion was completely abolished. However, this effect could be fully restored by pretreatment of the islets with the AT1-receptor antagonist, losartan. In addition, the AT1-receptors were specifically localised to the islet beta cells as evidenced by double immunofluorescence staining. This further indicates that the inhibitory action of Ang II was mediated through the AT1-receptors, which were located in the beta cells of pancreatic islets. Previous studies reported that receptors for Ang II have also been identified in the animal pancreatic islets [8, 32], but at least in rat, they were localised preferentially to the surface of alpha- and delta-cells [32]. In the human pancreas positive cells for AT1-receptor seem, however, to be distributed to those cells stained with insulin [13] although the precise localisation of the AT1-receptors has yet to be established.
In contrast to the marked effects of Ang II on insulin release from mouse pancreatic islets that we observed, exposure of the rat pancreatic islets to Ang II at a similar dose showed no effects on insulin release in a previous study [33]. This discrepancy of results could be due not only to the species difference but also to the different techniques used in these studies. In this respect, we examined the influence of Ang II on glucose-stimulated (16.7 mmol/l glucose) insulin release, whereas the effect of Ang II on insulin release was studied at low glucose concentration (5.6 mmol/l) in a previous study [33]. The currently employed approach with studies of isolated islets excluded that the inhibitory action of Ang II on insulin release was solely attributable to its vasoconstrictor actions in the pancreatic islets [16]. The inhibitory effect of Ang II on glucose-stimulated insulin secretion from isolated islets was due, at least partly, to the decrease in (pro)insulin biosynthesis. In contrast, islet glucose oxidation rate was unaffected by Ang II, indicating that the suppressive action of Ang II on glucose-stimulated insulin secretion appears not to be the changes in glucose oxidation. Ang II-receptors could influence prostaglandin synthesis, which in turn could modulate insulin secretion [34, 35]. Whether the inhibitory action of Ang II on insulin release from isolated islets is also mediated by this mechanism remains to be determined.
Most interestingly, a decreased incidence of Type 2 diabetes mellitus has been reported in several large clinical studies of patients at high risk of cardiovascular events treated with either ACE-inhibitors [36, 37] or Ang II-receptor antagonists [38, 39]. The exact mechanism(s) for this protection by these RAS blockers against diabetic patients remains equivocal. However, these drugs have been shown in some studies to improve the secretion and action of insulin. Although the effects of ACE-inhibition on insulin sensitivity are not clear, the beneficial effects of ACE-inhibition for insulin release have been shown in many hypertensive patients [40, 41, 42]. Moreover, intravenous infusion of Ang II has suppressed both basal and especially glucose-stimulated insulin secretion in humans [43]. Based on our findings, it is plausible to speculate that Ang II induced a dose-dependent inhibition of glucose-stimulated insulin release; this could be due to a decrease in (pro)insulin biosynthesis, which was mediated through the AT1-receptors located in islet beta cells. On the other hand, losartan per se had no effects on glucose-stimulated insulin release from isolated islets in our study. It is not known whether the islet angiotensin-generating system can change or become up-regulated during some disease states, e.g. Type 2 diabetes and hypertension. In such cases, the potential involvement of Ang II and its new regulatory pathway in islet insulin secretion may clarify the mechanism of RAS inhibition for the protection of Type 2 diabetes.
Islet-cell transplantation provides a possibility to accomplish permanent normoglycaemia in patients with Type 1 diabetes mellitus [44]. However, its clinical application is severely restricted by the requirement of considerable number of islet cells [45, 46], which might be due partly to a lack of proper engraftment thus leading to early graft failure [47]. A chronically low oxygen tension and a markedly decreased blood perfusion have been observed in transplanted islets [19, 20, 21]. Both chronic hypoxia and inflammatory conditions have activated and up-regulated RAS components in the pancreas [17, 27, 48]. In the present study, the expression of AT 1 -receptors, in contrast to the expression of AT 2 -receptors, ACE and Ao, was found markedly up-regulated both at the mRNA and protein levels in transplanted islets, as shown by real-time RT-PCR and western blot analysis, respectively. This observation is consistent with the enhanced vascular sensitivity to Ang II, when compared to endogenous islets, as recorded in islet transplants [49].
In conclusion, the study gives evidence for the presence of a local angiotensin-generating system in the islets. This system provides an inhibitory role for locally produced Ang II of glucose-stimulated insulin secretion, an effect mediated by the AT1-receptors located on the surface of the islet beta cells.
Abbreviations
- Ang II:
-
Angiotensin II
- AT1 :
-
angiotensin II receptor type 1
- AT2 :
-
angiotensin II receptor type 2
- Ao:
-
angiotensinogen
- RAS:
-
Renin-angiotensin system
- KRBB:
-
Krebs-Ringer bicarbonate buffer
References
De Gasparo M, Catt KJ, Inagami T, Wright JW, Unger H (2000) The angiotensin II receptors. Pharmacol Rev 52:415–472
Campell DJ (2003) The renin-angiotensin system and the kallikrein-kinin systems. Int J Biochem Cell Biol 35:784–791
Campbell DJ, Habener JF (1986) Angiotensinogen gene is expressed and differentially regulated in multiple tissues of the rat. J Clin Invest 78:31–39
Campbell DJ (1987) Circulating and tissue angiotensin systems. J Clin Invest 79:1–6
Phillips MI, Speakman EA, Kimura B (1993) Levels of angiotensin and molecular biology of the tissue renin-angiotensin systems. Regul Pept 43:1–20
Wang Y, Yamaguchi T, Francosaenz R, Mulrow PJ (1992) Regulation of renin gene expression in rat adrenal zona glomerulosa cells. Hypertension 20:766–781
Leung PS, Sernia C (2003) The renin-angiotensin system and male reproduction: new functions for old hormones. J Mol Endocrinol 30:263–270
Leung PS, Carlsson PO (2001) Tissue renin-angiotensin system: its expression, localization, regulation and potential role in the pancreas. J Mol Endocrinol 26:155–164
Chappell MC, Milsted A, Diz DI, Brosnihan KB, Ferrario CM (1991) Evidence for an intrinsic angiotensin system in the canine pancreas. J Hypertens 9:751–759
Leung PS, Chan HC, Fu LXM, Wong PYD (1997) Localization of angiotensin II receptor subtypes AT1 and AT2 in the pancreas of rodents. J Endocrinol 153:269–274
Leung PS, Chan WP, Wong TP, Sernia C (1999) Expression and localization of rennin-angiotensin system in the rat pancreas. J Endocrinol 160:13–19
Leung PS, Chan HC, Wong PYD (1998) Immunohistochemical localization of angiotensin II in the mouse pancreas. Histochem J 30:21–25
Tahmasebi M, Puddefoot JR, Inwang ER, Vinson GP (1999) The tissue rennin-angiotensin system in human pancreas. J Endocrinol 161:317–322
Leung PS, Chappell MC (2003) A local pancreatic renin-angiotensin system: endocrine and exocrine roles. Int J Biochem Cell Biol 35:838–846
Fink AS, Wang Y, Mendez T et al. (2002) Angiotensin II evokes calcium-mediated signaling events in isolated dog pancreatic epithelial cells. Pancreas 25:290–295
Carlsson PO, Berne C, Jansson L (1998) Angiotensin II and the endocrine pancreas: effects on islet blood flow and insulin secretion in rats. Diabetologia 41:127–133
Chan WP, Fung ML, Nobiling R, Leung PS (2000) Activation of local rennin-angiotensin system by chronic hypoxia. Mol Cell Endocrinol 160:107–114
Ip SP, Chan WY, Leung PS (2002) Effects of chronic hypoxia on the circulating and pancreatic rennin-angiotensin system. Pancreas 25:296–300
Carlsson PO, Liss P, Andersson A, Jansson L (1998) Measurements of oxygen tension in native and transplanted rat pancreatic islets. Diabetes 47:1027–1032
Carlsson PO, Palm F, Andersson A, Liss P (2001) Markedly decreased oxygen tension in transplanted rat pancreatic islets irrespective of the implantation site. Diabetes 50:489–495
Carlsson PO, Palm F, Andersson A, Liss P (2000) Chronically decreased oxygen tension in rat pancreatic islets transplanted under the kidney capsule. Transplantation 69:761–766
Andersson A (1978) Isolated mouse pancreatic islets in culture: effects of serum and different culture media on the insulin production of the islets. Diabetologia 14:397–404
Wang Z, Gleichmann H (1998) GLUT2 in pancreatic islets: crucial target molecule in diabetes induced with multiple low doses of streptozotocin in mice. Diabetes 47:50–56
Andersson A, Sandler S (1983) Viability tests of cryopreserved endocrine pancreatic cells. Cryobiology 20:161–168
Halban PA, Wollheim CB, Blondel B, Renold AE (1980) Long-term exposure of isolated pancreatic islets to mannoheptulose: Evidence for insulin degradation in the β-cell. Biochem Pharmacol 29:2625–2633
Carlsson PO, Palm F, Mattsson G (2002) Low revascularization of experimentally transplanted human pancreatic islets. J Clin Endcorinol Metab 87:5418–5423
Leung PS, Chan WP, Nobiling R (2000) Regulated expression of pancreatic rennin-angiotensin system in experimental pancreatitis. Mol Cell Endocrinol 166:121–128
Lam SY, Leung PS (2002) A locally generated angiotensin system in rat carotid body. Regul Pept 107:97–103
Fung ML, Lam SY, Dong X, Chen Y, Leung PS (2002) Postnatal hypoxemia increases angiotensin II sensitivity and up-regulates AT1a receptors in rat carotid body chemoreceptors. J Endocrinol 173:305–313
Winer J, Jung KS, Shackel I, Williams PM (1999) Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem 270:41–49
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408
Ghiani BU, Masini MA (1995) Angiotensin II binding sites in the rat pancreas and their modulation after sodium loading and depletion. Comp Biochem Physiol 111A:439–444
Dunning BE, Moltz JH, Fawcett CP (1984) Actions of neurohypophysial peptides on pancreatic hormone release. Am J Physiol 246:108–114
Jaiswal N, Diz DI, Taillant EA, Kosla MC, Ferrario CM (1991) Characterization of angiotensin receptors mediating prostaglandin synthesis in C6 glioma cells. Am J Physiol 260:1000–1006
Kelly KL, Laychock SG (1981) Prostaglandin synthesis and metabolism in isolated pancreatic islets of the rat. Prostaglandins 21:756–769
Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G (2000) The Heart Outcomes Prevention Evaluation Study, effects of an angiotensin-converting enzyme inhibitor ramipril on cardiovascular events in high-risk patients. N Engl J Med 342:145–153
Hansson L, Lindholm DH, Niskanen L, Lanke J, Hedner T, Niklason A (1999) Effects of angiotensin-converting-enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPP) randomised trial. Lancet 353:611–616
Dahlöf B, Devereux RB, Kjeldsen SE et al. (2002) Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoints reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 359:995–1003
Califf RM (2003) Insulin resistance: a global epidemic in need of effective therapies. Eur Heart J 5 [Suppl C]:C13–C18
Pollare T, Lithell H, Berne C (1989) A comparison of the effects of hydrochlorothiazide and captopril on glucose and lipid metabolism in patients with hypertension. N Engl J Med 321:868–873
Hänni A, Andersson PE, Lind L, Berne C, Lithell H (1994) Electrolyte changes and metabolic effects of lisinopril/bendrofluazide treatment. Results from a randomised, double-blind study with parallel groups. Am J Hypertens 7:615–622
Santoro D, Natali A, Palombo C et al. (1992) Effects of chronic angiotensin converting enzyme inhibition on glucose tolerance and insulin sensitivity in essential hypertension. Hypertension 20:181–191
Fliser D, Kohl B, Hartung R, Ritz E (1993) Suppressor doses of angiotensin II increases insulin sensitivity in healthy volunteers. J Hypertens 11:983–988
Kenyon N, Alejandro R, Mintz D, Ricordi C (1996) Islet cell transplantation: beyond the paradigms. Diabetes Metab Rev 12:361–372
Ryan EA, Lakey JRT, Rajotte RV et al. (2001) Clinical outcomes and insulin secretion after islet transplantation with the Edmonton protocol. Diabetes 50:710–719
Brendel M, Hering B, Schultz A, Bretzel R (2001) Newsletter no. 9. Int Islet Transpl Reg 8:1–20
Jansson L, Carlsson P-O (2002) Graft vascular function after transplantation of pancreatic islets. Diabetologia 45:749–763
Ip SP, Kwan PC, Williams CH, Pang S, Hooper NM, Leung PS (2003) Changes of angiotensin-converting enzyme activity in the pancreas of chronic hypoxia and acute pancreatitis. Int J Biochem Cell Biol 35:944–964
Olsson R, Jansson L, Andersson A, Carlsson PO (2000) Local blood flow regulation in transplanted rat pancreatic islets: influence of adenosine, angiotensin II and nitric oxide inhibition. Transplantation 70:280–287
Acknowledgements
The study was fully supported by the Mainline Research Scheme from the Chinese University of Hong Kong (MR00/008) and by the Competitive Earmarked Research grant from the Research Grants Council of Hong Kong (project no. CUHK 4075/00M and 4116/01M). Losartan was kindly provided by Merck & Co., N.J., USA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lau, T., Carlsson, PO. & Leung, P.S. Evidence for a local angiotensin-generating system and dose-dependent inhibition of glucose-stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia 47, 240–248 (2004). https://doi.org/10.1007/s00125-003-1295-1
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1295-1