
Abstract
Profiling of RNA binding protein targets in vivo provides critical insights into the mechanistic roles they play in regulating RNA processing. The enhanced crosslinking and immunoprecipitation (eCLIP) methodology provides a framework for robust, reproducible identification of transcriptome-wide protein-RNA interactions, with dramatically improved efficiency over previous methods. Here we provide a step-by-step description of the eCLIP method, along with insights into optimal performance of critical steps in the protocol. In particular, we describe improvements to the adaptor strategy that enables single-end enhanced CLIP (seCLIP), which removes the requirement for paired-end sequencing of eCLIP libraries. Further, we describe the observation of contaminating RNA present in standard nitrocellulose membrane suppliers, and present options with significantly reduced contamination for sensitive applications. These notes further refine the eCLIP methodology, simplifying robust RNA binding protein studies for all users.
The original version of this chapter was revised. The erratum to this chapter is available at: DOI 10.1007/978-1-4939-7204-3_19
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others



Key words
1 Introduction
RNA processing has been shown to play pivotal roles in shaping the cellular landscape through regulation of both protein-coding RNAs as well as regulation of processing and function of multiple classes of noncoding RNAs including long intergenic noncoding RNAs (lincRNAs) and small RNAs including microRNAs and piRNAs among others [1,2,3]. These regulatory steps have been shown to play critical roles across a variety of developmental stages, and misregulation of RNA processing has been implicated in many human diseases, including cancer [2, 3]. Orchestrating these RNA processing and regulatory roles are RNA binding proteins (RBPs), which play a variety of roles including controlling alternative splicing of mRNA transcripts, targeting RNAs to specific organelles or subcellular localizations with the cell, controlling RNA stability and turnover, and defining the timing and rate of translation [4]. Recent studies have indicated that there are over 1500 RBPs, and this number is continuing to expand with additional studies [5]. Despite these known important roles, however, detailed studies to describe the targets of and regulatory mechanisms have largely focused on a small set of RBPs, with the majority of RBPs remaining poorly characterized.
Recent advancements in next-generation sequencing have made it possible to study RBPs and their RNA targets in an unbiased and transcriptome-wide manner [6, 7]. Building upon early RNA ImmunoPrecipitation (RIP) approaches that identified protein binding to entire transcripts, CrossLinking and ImmunoPrecipitation (CLIP) enabled high-resolution profiling of binding sites [8]. In CLIP, RNA-protein interactions are stabilized via ultraviolet crosslinking, a desired protein is immunoprecipitated using a factor-specific antibody, and associated RNA is isolated and converted into DNA library suitable for high-throughput sequencing [8]. Various modifications of CLIP have since been described, including the use of photoactivatable nucleoside analogs (PNAs) to improve crosslinking efficiency (PAR-CLIP) [9] and computational and experimental methods to identify binding with single-nucleotide resolution. Proteinase K treatment of UV-crosslinked protein-RNA complexes leaves at least one amino acid covalently crosslinked to its associated ribonucleotide. Reverse transcriptase enzymes can create deletions at these positions [10] or, more often, terminate elongation due to the inability to read through this coupling, leading to a substantial fraction of cDNA fragments that terminate at the position of crosslinking. By incorporating a circular ligation step, iCLIP positions this crosslinking site at the start of the sequencing reads to enable identification of binding sites with single-nucleotide resolution [11]. However, widespread usage of these methods has been limited by the low efficiency of converting RNA molecules into adapter-ligated library , which leads to high experimental failure rates and high wasted sequencing (often >90% of reads) due to the presence of substantial PCR duplication [12].
We recently described enhanced CLIP (eCLIP ), which incorporated high-efficiency enzymatic steps to achieve thousand-fold improved library efficiency [12]. The improved efficiency dramatically decreases experimental failure rates and PCR duplication, and enabled quantitative comparison with paired size-matched input to remove common CLIP artifacts. Here, we describe a detailed protocol for seCLIP, a simplified, single-end version of the eCLIP methodology, as well as assorted notes for critical handling steps. By ligating an adapter at the 3′ end of the cDNA fragment, eCLIP (similar to iCLIP) utilizes this termination to enrich for read pileups at these sites. Due to the adapter strategy used in our initial eCLIP procedure that positioned the I7 adaptor at this site, eCLIP initially required paired-end sequencing to obtain these crosslink sites at the 5′ end of the second read, as well as the unique molecular identifier (UMI, or random-mer) used for PCR duplicate identification. In this protocol, we describe an altered adapter strategy to enable single-end sequencing for eCLIP experiments.
Additionally, during eCLIP experiments on cell types with low total RNA quantity, we observed that a substantial fraction (in some cases more than half) of sequenced reads did not map to the human genome and instead mapped to a single bacterial contamination. We trace this source of contamination to manufacturer-supplied nitrocellulose membranes, and describe alternate sources for this material that alleviate this contamination and a method to assay membranes for eCLIP suitability. With a new 3′ linker ligation strategy that allows for single-end sequencing, and the alleviation of contamination found commonly in major suppliers, seCLIP brings modifications to eCLIP that will allow for more cost-efficient sequencing as well as paving the way for future low-input RNA as starting material for CLIP-seq experiments.
2 Materials
2.1 Crosslinking of Cultured Cells
-
1.
1× DPBS.
-
2.
254 nM UV crosslinker.
-
3.
Cell scraper.
-
4.
Liquid Nitrogen.
2.2 seCLIP
-
1.
Lysis buffer: 50 mM Tris–HCl pH 7.4, 100 mM NaCl, 1% NP-40 (Igepal CA630), 0.1% SDS, 0.5% sodium deoxycholate (protect from light), 1:200 Protease Inhibitor Cocktail III (add fresh), in RNase /DNase-free H2O.
-
2.
Protease Inhibitor Cocktail III.
-
3.
DNase.
-
4.
RNase I.
-
5.
RNase Inhibitor.
-
6.
Dynabeads M-280 sheep anti-rabbit or Protein A/G magnetic beads.
-
7.
High salt wash buffer: 50 mM Tris–HCl pH 7.4, 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate (protect from light), in RNase/DNase-free H2O.
-
8.
Wash buffer: 20 mM Tris–HCl pH 7.4, 10 mM MgCl2, 0.2% Tween-20, in RNase /DNase-free H2O.
-
9.
1× TAP Buffer: 10 mM Tris pH 7.5, 5 mM MgCl2,100 mM KCl, 0.02% Triton X-100, in RNase/DNase-free H2O.
-
10.
Thermosensitive Alkaline Phosphatase (TAP) (1 unit/μL).
-
11.
5× PNK pH 6.5 buffer: 350 mM Tris–HCl pH 6.5, 50 mM MgCl2, in RNase/DNase-free H2O.
-
12.
0.1 M DTT.
-
13.
T4 PNK.
-
14.
1× RNA Ligase Buffer: 50 mM Tris–HCl pH 7.5, 10 mM MgCl2, in RNase/DNase-free H2O.
-
15.
10× Ligase Buffer without DTT.
-
16.
0.1 M ATP.
-
17.
100% DMSO.
-
18.
50% PEG 8000.
-
19.
T4 RNA ligase 1 high concentration.
-
20.
4–12% Bis-Tris Gel.
-
21.
NuPAGE 4× LDS Sample Buffer.
-
22.
NuPAGE MOPS SDS Running Buffer 20×.
-
23.
NuPAGE Transfer Buffer 20×.
-
24.
PVDF membrane.
-
25.
Nitrocellulose membrane (see Note 4 ):
(a) iBlot 2 Transfer Stacks | ThermoFisher | IB23001 | lot #2NR26016-01 |
or
(b) Amersham Protran Premium | GE | 1060008 | lot #G9931040 |
-
26.
5% milk + TBST (1× TBS pH 7.4 + 0.05% Tween-20).
-
27.
Rabbit TrueBlot HRP secondary antibody.
-
28.
ECL Western Blotting detection assay.
-
29.
Proteinase K .
-
30.
Urea.
-
31.
Acid Phenol/Chloroform/Isoamyalcohol pH 4.5.
-
32.
Phase lock heavy 2 mL Tubes.
-
33.
100% Ethanol.
-
34.
RNA Clean & Concentrator-5 Kit.
-
35.
Dynabeads MyOne Silane.
-
36.
RLT Buffer.
-
37.
5 M NaCl.
-
38.
10× Ligase Buffer with DTT.
-
39.
10× AffinityScript reverse transcriptase buffer.
-
40.
AffinityScript reverse transcriptase.
-
41.
dNTPs (25 mM each).
-
42.
Exo-SAP-IT.
-
43.
0.5 M EDTA.
-
44.
1 M NaOH.
-
45.
1 M HCl.
-
46.
5 mM Tris–HCl pH 7.5.
-
47.
10 mM Tris–HCl pH 7.5.
-
48.
Q5 or other high fidelity PCR Master Mix.
-
49.
qPCR Master Mix.
-
50.
Agencourt AMPure XP beads.
-
51.
MinElute gel purification Kit.
-
52.
D1000 DNA tape/reagent.
2.3 Contamination Assay
-
1.
TRIzol® Reagent.
-
2.
TRIzol® LS Reagent.
-
3.
SuperScript II (200 unit/μL).
2.4 Primer Sequences
seCLIP:
-
1.
InvRiL19: /5Phos/rArGrArUrCrGrGrArArGrArGrCrArCrArCrGrUrC/3SpC3/
(Order 100 nmole RNA oligo, standard desalting; storage stock 200 μM; working stock 40 μM; final concentration 1 μM (input), 4 μM (CLIP)).
-
2.
InvRand3Tr3: /5Phos/NNNNNNNNNNAGATCGGAAGAGCGTCGTGT/3SpC3/
(Order 100 nmole DNA oligo, standard desalting; storage stock 200 μM; working stock 80 μM; final concentration 3 μM).
-
3.
InvAR17: CAGACGTGTGCTCTTCCGA (25 nmole DNA oligo, standard desalting; storage stock 200 μM; working stock 20 μM; final concentration 0.5 μM).
-
4.
D5x_qPCR: AATGATACGGCGACCACCGAGATCTACACTATAGCCTACACTCTTTCCCTACACGACGCTCTTCCGATCT.
-
5.
D7x_qPCR: CAAGCAGAAGACGGCATACGAGATCGAGTAATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC.
XBB1 contamination primers:
-
1.
XBB1_qPCR _F: GAGGCGGCAAATATCCTGTG.
-
2.
XBB1_qPCR_R: GTTTCACTTCCCCTCGTTCG.
3 Methods
See Fig. 1 for schematic of eCLIP methodology.

Schematic of seCLIP method. (a) Crosslinking of cultured cells (Subheadings 3.1.1 and 3.1.2). (b) Lysis of crosslinked cells (Subheading 3.2.1). (c) RNA fragmentation with RNase (Subheading 3.2.2). (d) Immunoprecipitation of RBP -RNA complexes (Subheadings 3.3.1–3.3.5). (e) Dephosphorylation of RNA fragments and ligation of 3′ RNA adapter (Subheading 3.5.1). (f) Polyacrylamide gel electrophoresis and membrane transfer (Subheadings 3.6.1–3.6.5). (g) Mince preparative membrane into ~2 mm squares (Subheadings 3.6.6) (h) RNA isolation from membrane (Subheadings 3.6.7 and 3.6.8). (i) Dephosphorylation of RNA fragments and ligation of 3′ RNA adapter for input samples (Subheadings 3.7.1–3.8.3). (j) Reverse transcription of RNA (Subheadings 3.9.1 and 3.9.2). (k) cDNA cleanup (removal of excess primers and RNA) (Subheading 3.9.3). (l) Ligation of 3′ DNA adapter (on-bead) and cleanup (Subheadings 3.10.1–3.10.3). (m) PCR amplification of cDNA library and cleanup (Subheadings 3.11.2–3.12.2). (n) Final Structure of eCLIP library fragment. The unique molecular identifier or random-mer is shown in green and abbreviated as UMI
3.1 Crosslinking of Cultured Cells
3.1.1 Preparation of Cultured Cells for Crosslinking
-
Aspirate the spent media and wash the plate with 1× DPBS (~15 mL for a 15 cm plate) at room temperature. For suspension cells, pellet cells to remove spent media, and wash once with 1× DPBS.
-
Aspirate the supernatant from previous, and add enough 1× DPBS (~5 mL for a 15 cm plate) to cover the plate. For suspension cells, resuspend in 1× DPBS to a cell density of 2 × 107 cells per mL, place 3 mL of cell suspension in 1× DPBS on a 10 cm plate.
3.1.2 Crosslinking of Cultured Cells
-
Place the tissue culture plate on ice or cooling block, and place the entire apparatus into the UV crosslinker, making sure the plate is leveled. Remove lid prior to crosslinking.
-
Crosslink the cells with an energy setting of 400 mJ/Cm2.
-
While keeping the cells on ice, use a cell scraper to scrape the plate. Transfer the cells to a 50 mL conical tube. Wash the plate with 10 mL of 1× DPBS and add the wash to the 50 mL conical tube. Gently resuspend the cell until a homogenous mixture is obtained.
-
Spin cells down at 200 × g for 5 min at room temperature.
-
Aspirate the supernatant. Resuspend in the desired amount for flash freezing, typically 2 × 107 cells per mL.
-
Transfer desired amount into 1.5 mL epi-tubes, and then spin down at 200 × g for 5 min at room temperature. Aspirate the supernatant and freeze by submerging the epi-tubes completely in liquid nitrogen. Store at −80 °C.
3.2 Cell Lysis and RNA Fragmentation
3.2.1 Lyse Cells
-
Add 1 mL cold lysis buffer + 5.5 μL 200× Protease Inhibitor Cocktail III to each pellet (see Note 1 ).
-
Pipette to resuspend, incubate for 15 min on ice.
-
At this time, begin antibody coupling (Subheading 3.3.1).
-
3.2.2 RNase Treat Lysate
-
Sonicate in Bioruptor at “low” setting, 30 s on / 30 s off for 5 min at 4 °C.
-
Add 2 μL DNase to lysate.
-
Dilute RNase I 1:25 in 1× PBS (prechilled to 4 °C).
-
Add 10 μL diluted RNase I to lysate, mix and immediately proceed to the next step.
-
Incubate in Thermomixer at 1200 rpm, 37 °C for 5 min.
-
Place on ice, add 11 μL Murine RNase Inhibitor and pipette mix (see Note 1 ).
-
Centrifuge 15,000 × g, 4 °C for 15 min → Transfer the supernatant to a new tube and discard pellet.
3.3 Capture RBP -RNA Complexes on Beads
3.3.1 Couple Antibody to Magnetic Beads (Start During Subheading 3.2.1)
-
Use 125 μL beads + 10 μg antibody per sample.
3.3.2 Prewash Beads
-
Magnetically separate beads, remove the supernatant.
-
Wash beads 2× in 500 μL cold lysis buffer.
-
Resuspend beads in 100 μL cold lysis buffer.
3.3.3 Bind Antibody
-
Add 10 μg of antibody to 100 μL washed beads.
-
Rotate at room temperature for 45 min.
3.3.4 Capture RBP -RNA Complexes on Beads
-
Wash antibody beads 2× in 500 μL cold lysis buffer.
-
Add the supernatant from Subheading 3.2.2 to washed antibody beads.
-
Rotate at 4 °C overnight
3.3.5 Remove Input Samples
-
To a new tube, take 20 μL (2%) of total beads + lysate sample for Preparative gel, store at 4 °C.
-
To a new tube, take 20 μL (2%) of total beads + lysate sample for Imaging gel; store at 4 °C.
3.4 Immunoprecipitation Washes and RNA Dephosphorylation
3.4.1 Wash Beads (Pre-chill All Wash Buffers to 4 °C) (See Note 2 )
-
Magnetically separate beads, remove the supernatant (or store for western if desired).
-
Wash 2× with 900 μL High salt wash buffer, then 1× with 500 μL Wash buffer. Remove the supernatant.
-
Add 500 μL Wash buffer, mix, separate on a magnet, add 500 μL 1× TAP buffer, mix, remove the supernatant. Wash 1× with 500 μL 1× TAP buffer, remove the supernatant.
3.4.2 TAP Treat (on-Bead)
-
Prepare TAP master mix on ice (100 μL per sample):
H2O | 79 μL |
10× TAP buffer | 10 μL |
Murine RNase Inhibitor | 2 μL |
DNase | 1 μL |
TAP enzyme | 8 μL |
-
Add 100 μL TAP mix to each sample, incubate in Thermomixer at 1200 rpm, 37 °C for 15 min.
3.4.3 PNK Treat (On-Bead)
-
Prepare PNK master mix on ice (300 μL per sample):
H2O | 224 μL |
5× PNK pH 6.5 buffer | 60 μL |
0.1 M DTT | 3 μL |
Murine RNase Inhibitor | 5 μL |
DNase | 1 μL |
T4 PNK enzyme | 7 μL |
-
Add 300 μL PNK mix to each sample, incubate in Thermomixer at 1200 rpm, 37 °C for 20 min.
3.4.4 Wash Beads (Prechill Buffers to 4 °C)
-
Magnetically separate beads, remove the supernatant.
-
Wash with 1× 500 μL Wash buffer, remove the supernatant.
-
Add 500 μL Wash buffer, mix, add 500 μL High salt wash buffer, mix, remove the supernatant.
-
Add 500 μL High salt wash buffer, mix, add 500 μL Wash buffer, mix, remove the supernatant.
-
Wash 1× with 500 μL Wash buffer, remove the supernatant.
-
Add 500 μL Wash buffer, mix, add 300 μL 1× RNA Ligase buffer (no DTT), mix, remove the supernatant.
-
Repeat wash 2× with 300 μL 1× RNA Ligase buffer (no DTT), carefully remove all remaining supernatant.
3.5 Ligate 3′ RNA Adapter (On-Bead)
3.5.1 Prepare RNA Adapter Ligation Master Mix
-
Prepare on ice; 25 μL per sample:
H2O | 9 μL |
10× Ligase buffer (no DTT) | 3 μL |
0.1 M ATP | 0.3 μL |
100% DMSO | 0.8 μL |
50% PEG 8000 | 9 μL |
Murine RNase Inhibitor | 0.4 μL |
High concentration T4 RNA Ligase | 2.5 μL |
-
Mix carefully by pipetting (do not vortex) (see Note 3 ).
3.5.2 Perform RNA Adapter Ligation
-
Add 25 μL RNA adapter ligation master mix to each sample.
-
Add 2.5 μL InvRiL19 RNA adapter to each sample.
-
Incubate at room temperature for 75 min; flick to mix every ~10 min.
3.6 Western Blotting and RNA Isolation from Membrane
3.6.1 Wash Beads (Prechill Buffers to 4 °C)
-
Add 500 μL Wash buffer, magnetically separate, remove the supernatant.
-
Add 500 μL Wash buffer, mix, add 500 μL High salt wash buffer, mix, remove the supernatant.
-
Wash 1× with 500 μL High salt wash buffer, remove the supernatant.
-
Add 500 μL High salt wash buffer, mix, add 500 μL Wash buffer, mix, remove the supernatant.
-
Repeat wash 2× with 500 μL Wash buffer.
3.6.2 Prepare Samples for Gel Loading
-
Preparative gel—used for membrane transfer and RNA isolation.
-
Imaging gel—for IP-western blot imaging of IP success.
-
Remove supernatant, add 100 μL cold Wash buffer, resuspend beads well.
-
Move 20 μL (20% of sample) to new tube #1 for Imaging gel IP samples.
-
Magnetically separate, remove the supernatant.
-
Resuspend sample in 20 μL cold Wash buffer = Preparative gel IP samples.
-
Thaw on ice Preparative and Imaging input samples saved at Subheading 3.3.5.
-
Prepare each sample mix:
IP or input sample | 20 μL |
4× NuPAGE buffer | 7.5 μL |
1 M DTT | 3.0 μL |
-
Denature all samples in Thermomixer (1200 rpm, 70 °C for 10 min), and then cool on ice >1 min.
-
For all samples, magnetically separate and only load supernatant (IP AND Inputs have beads).
3.6.3 Load and Run Gels
-
Load Preparative gel (4–12% Bis-Tris, 10-well, 1.5 mm) with (M) prestained markers and (m) diluted prestained marker (2 μL marker +2 μL 4× NuPAGE buffer +6 μL Wash Buffer) as follows for two experiments (A and B), leaving marker-only lanes between samples:
1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|
M | A-Input | (m) | A-IP | (m) | B-Input | (m) | B-IP | M | (m) |
-
Load: 30 μL (all) volume for all preparative samples (input and IP).
-
Load Imaging gel (4–12% Bis-Tris, 10 or 12-well, 1.5 mm).
-
Load 15 μL for all cold samples, save remaining volume at −20 °C as backup.
-
-
Run at 150 V in 1× MOPS running buffer for 75 min or until dye front is at the bottom.
3.6.4 Transfer to Membranes
-
Prepare transfer: (use 4 °C transfer buffer)
-
Imaging gel: Use PVDF membrane, activate with Methanol, and then move to transfer buffer.
-
Preparative gel: Use Nitrocellulose membrane (see Note 4 ), presoak in transfer buffer.
-
Assemble transfer stacks, from bottom to top (black side (negative) of stack holder on bottom):
(negative)1× sponge – 2× Whatman – gel – membrane – 2× Whatman – 1× sponge (positive).
-
-
Transfer:
-
Overnight at 30 V (preferred) or 2 h at 200 mA.
-
After removing Preparative membrane, rinse quickly once with sterile 1× PBS, then wrap in Saran wrap and store at −20 °C until Subheading 3.6.6.
-
3.6.5 Develop Imaging Membrane
-
Block in 5% milk in TBST at room temperature for 30 min.
-
Probe with primary antibody at appropriate concentration (typically 0.2 μg/mL) in 5% milk in TBST, at room temperature for 1 h.
-
Wash 3× with TBST, 5 min.
-
Probe with secondary antibody: 1:4000 Rabbit TrueBlot HRP in 5% milk in TBST, incubate at room temperature for 1–3 h.
-
Wash 3× with TBST, 5 min.
-
Develop with ECL, 30 s to 5 min, image with standard western blot film.
3.6.6 Cut Preparative Membrane (Cut Bands Based on Western Image)
-
Place preparative membrane on clean glass/metal surface.
-
Using a fresh razor blade, cut desired lane from the RBP band to 75 kDa above.
-
Slice membrane piece into ~1–2 mm slices.
-
Transfer slices to Eppendorf tube—place tube on ice if doing many samples.
3.6.7 Release RNA from Membrane
ProK mix on ice, 200 μL per sample: • PK buffer: 160 μL • Proteinase K : 40 μL | Urea/PK buffer • Dissolve 420 mg Urea in 500 μL PK buffer, then add PK buffer to a final volume of 1 mL |
-
Add 200 μL ProK mix to membrane slices, incubate in Thermomixer (1200 rpm, 37 °C for 20 min).
-
Add 200 μL Urea/PK buffer to samples, mix, incubate in Thermomixer (1200 rpm, 37 °C for 20 min).
3.6.8 Purify RNA
-
Add 400 μL acid phenol/chloroform/isoamyl alcohol (pH 4.5), mix well, incubate in Thermomixer at 1200 rpm, 37 °C for 5 min.
-
Transfer all except membrane slices to Phaselock gel (Heavy) tube, incubate in Thermomixer at 1200 rpm, 37 °C for 5 min.
-
Centrifuge at 13,000 × g for 15 min at room temperature.
-
Transfer the aqueous (top) layer to a new 15 mL (or at least 3 mL volume) conical tube.
-
Add 2 volumes RNA binding buffer (typically 2× ~400 = 800 μL).
-
Add equal volume 100% ethanol and mix (typically ~1200 μL).
-
Transfer 750 μL of mixed sample to Zymo-Spin column.
-
Centrifuge for 30 s and discard flow-through.
-
Repeat spins by reloading additional 750 μL volume until all sample has been spun through column.
-
Add 400 μL RNA Prep Buffer, centrifuge for 30 s, discard flow-through.
-
Add 700 μL RNA Wash Buffer, centrifuge for 30 s, discard flow-through.
-
Add 400 μL RNA Wash Buffer, centrifuge for 30 s, discard flow-through.
-
Centrifuge for additional 2 min.
-
Transfer column to a new 1.5 mL tube (avoid getting wash buffer on column).
-
Add 10 μL H2O to column, let it sit for 1 min, centrifuge for 30 s.
-
Store CLIP samples at −80 °C until Subheading 3.9 (avoid multiple freeze-thaw cycles).
3.7 Dephosphorylation of Input RNA
3.7.1 TAP Treat Input RNA
-
Prepare TAP master mix
– H2O | 10 μL |
– 10× TAP buffer | 2.5 μL |
– RNase Inhibitor | 0.5 μL |
– TAP enzyme | 2.5 μL |
-
To INPUT samples ONLY, add 15.5 μL TAP master mix.
-
Mix and incubate in Thermomixer at 1200 rpm, 37 °C for 15 min.
3.7.2 PNK Treat Input RNA
-
Prepare PNK master mix (75 μL per sample).
– H2O | 45 μL |
– 5× PNK pH 6.5 buffer | 20 μL |
– 0.1 M DTT | 1 μL |
– DNase | 1 μL |
– Murine RNase Inhibitor | 1 μL |
– T4 PNK enzyme | 7 μL |
-
Add 75 μL to samples and mix. Incubate in Thermomixer at 1200 rpm, 37 °C for 20 min.
3.7.3 Silane Cleanup Input RNA
-
Prepare beads:
-
Magnetically separate 20 μL MyONE Silane beads per sample, remove the supernatant.
-
Wash 1× with 900 μL RLT buffer.
-
Resuspend beads in 300 μL RLT buffer per sample.
-
-
Bind RNA:
-
Add beads in 300 μL RLT buffer to sample, mix.
-
Add 10 μL 5 M NaCl.
-
Add 615 μL 100% EtOH.
-
Mix, rotate at room temperature, 15 min.
-
-
Wash beads:
-
Magnetically separate, remove the supernatant.
-
Add 1 mL 75% EtOH, pipette resuspend and move suspension to new tube.
-
After 30 s, magnetically separate, remove the supernatant, and wash 2× with 75% EtOH (30 s).
-
Magnetically separate, remove residual liquid with fine tip → air-dry 5 min.
-
-
Elute RNA:
-
Resuspend in 10 μL H 2 O, let it sit for 5 min.
-
Magnetically separate and transfer 5 μL of supernatant to a new tube (for 3′ RNA adapter ligation below). Transfer the remainder of the supernatant to a new tube and store at −20 °C (as a backup input RNA sample).
-
3.8 3' RNA Adapter Ligation to Input RNA
3.8.1 Anneal Adapter
-
Take 5 μL of RNA (from above).
-
Add 1.5 μL 100% DMSO and 0.5 μL InvRiL19 adapter (see Note 3 ).
-
Incubate at 65 °C for 2 min. Place on ice >1 min.
3.8.2 Prepare Ligation Master Mix; 13.5 μL Per Sample
10× Ligase Buffer (with DTT) | 2.0 μL |
0.1 M ATP | 0.2 μL |
Murine RNase Inhibitor | 0.2 μL |
100% DMSO | 0.3 μL |
50% PEG 8000 | 8.0 μL |
RNA Ligase high conc | 1.3 μL |
H2O | 1.5 μL |
-
Add 13.5 μL to each sample, mix, incubate at room temperature for 75 min. Flick to mix every ~15 min.
3.8.3 Silane Cleanup Input RNA
-
Prepare beads:
-
Magnetically separate 20 μL MyONE Silane beads per sample, remove the supernatant.
-
Wash 1× with 900 μL RLT buffer and resuspend beads in 61.6 μL RLT buffer.
-
Bind RNA:
-
Add beads in 61.6 μL RLT buffer to sample, mix and add 61.6 μL 100% EtOH.
-
Pipette mix, leave pipette tip in a tube, pipette mix every ~3–5 min for 15 min.
-
-
Wash beads:
-
Magnetically separate, remove the supernatant.
-
Add 1 mL 75% EtOH, pipette resuspend and move to a new tube.
-
After 30 s, magnetically separate, remove the supernatant, and wash 2× with 75% EtOH (30 s).
-
Magnetically separate, remove residual liquid with fine tip → Air-dry 5 min.
-
-
Elute RNA:
-
Resuspend in 10 μL H 2 O, let it sit for 5 min.
-
Magnetically separate, transfer the supernatant to a new tube.
-
3.9 Reverse Transcribe RNA (All Clip and Input Samples), and Reaction Cleanup
3.9.1 Anneal Primer in 8-Well Strip Tubes
-
Mix 10 μL of RNA with 0.5 μL InvAR17 primer.
-
Heat at 65 °C for 2 min in preheated PCR block, place immediately on ice.
3.9.2 Prepare Reverse Transcription Master Mix on Ice; 10 μL Per Sample
– H2O | 4.0 μL |
– 10× AffinityScript Buffer | 2.0 μL |
– 0.1 M DTT | 2.0 μL |
– dNTPs (100 mM; 25 mM each) | 0.8 μL |
– Murine RNase Inhibitor | 0.3 μL |
– AffinityScript Enzyme | 0.9 μL |
-
Add 10 μL to each sample, mix, and incubate at 55 °C for 45 min in preheated PCR block.
3.9.3 Cleanup cDNA
-
Removal of excess primers
-
Add 3.5 μL ExoSAP-IT to each sample, vortex, spin down.
-
Incubate at 37 °C for 15 min on a PCR block.
-
Add 1 μL 0.5 M EDTA, pipette-mix.
-
-
RNA removal
-
Add 3 μL 1 M NaOH, pipette-mix.
-
Incubate at 70 °C for 12 min on a PCR block.
-
Add 3 μL 1 M HCl, pipette-mix.
3.9.3.1 Silane Cleanup cDNA
-
Prepare beads:
-
Magnetically separate 10 μL MyONE Silane beads per sample, remove the supernatant.
-
Wash 1× with 500 μL RLT buffer and resuspend beads in 93 μL RLT buffer.
-
-
Bind RNA:
-
Add beads in 93 μL RLT buffer to sample, mix and add 111.6 μL 100% EtOH.
-
Pipette mix, leave pipette tip in a tube, pipette mix twice, for 5 min.
-
-
Wash beads:
-
Magnetically separate, remove the supernatant.
-
Add 1 mL 80% EtOH, pipette resuspend and move to new tube.
-
After 30 s, magnetically separate, remove the supernatant, and wash 2× with 80% EtOH (30 s).
-
Magnetically separate, remove residual liquid with fine tip → air-dry 5 min.
-
-
Elute RNA:
-
Resuspend in 5 μL 5 mM Tris–HCl pH 7.5, let it sit for 5 min (do NOT remove from beads).
-
3.10 3' Linker Ligate cDNA (On-Bead), and Cleanup
3.10.1 Anneal Linker
-
Add 0.8 μL InvRand3Tr3 adapter (see Note 3 ).
-
Add 1 μL 100% DMSO.
-
Heat at 75 °C, 2 min, place immediately on ice for >1 min.
3.10.2 Prepare Ligation Master Mix on Ice; 12.8 μL Per Sample
– 10× RNA Ligase Buffer (with DTT) | 2.0 μL |
– 0.1 M ATP | 0.2 μL |
– 50% PEG 800 | 9.0 μL |
– High concentration T4 RNA Ligase | 0.5 μL |
– H2O | 1.1 μL |
-
Flick to mix, spin down, and add 12.8 μL to each sample: add master mix slowly with stirring; it needs to be homogeneous.
-
Add an additional 1 μL High concentration T4 RNA Ligase on the top of sample and pipette mix.
-
Incubate at room temperature overnight.
3.10.3 Silane Cleanup Linker-Ligated cDNA
-
Prepare beads:
-
Magnetically separate 5 μL MyONE Silane beads per sample, remove the supernatant.
-
Wash 1× with 500 μL RLT buffer.
-
Resuspend beads in 60 μL RLT buffer per sample.
-
-
Bind RNA:
-
Add beads in 60 μL RLT buffer to each sample, mix and add 60 μL 100% EtOH.
-
Pipette mix, incubate for 5 min at room temperature (pipette mix twice with same tips during incubation).
-
-
Wash beads:
-
Magnetically separate, remove the supernatant.
-
Add 1 mL 75% EtOH, pipette resuspend and move to new tube.
-
After 30 s, magnetically separate, remove the supernatant.
-
Wash 2× with 75% EtOH (30 s).
-
Magnetically separate, remove residual liquid with fine tip → Air-dry 5 min.
-
-
Elute RNA:
-
Resuspend in 27 μL 10 mM Tris–HCl pH 7.5, let it sit for 5 min.
-
Magnetically separate, transfer 25 μL sample to a new tube.
-
3.11 qPCR Quantify cDNA, PCR, and Reaction Cleanup
3.11.1 Prepare qPCR Master Mix; 9 μL Per Sample
– qPCR 2× master mix | 5.0 μL |
– H2O | 3.6 μL |
– qPCR primer mix | 0.4 μL (10 μM each qPCR-grade D5x/D7x mix) |
-
Mix, dispense into a 384-well qPCR plate.
-
Add 1 μL 1:10 diluted (in H 2 O) sample cDNA, seal, mix.
-
Run qPCR, note Cq values.
3.11.2 PCR Amplify cDNA
Prepare PCR on ice; 50 μL total per sample: • 2× PCR master mix: 25.0 μL • H2O: 7.5 μL • 20 μM forward primer (D50x): 2.5 μL • 20 μM reverse primer (D70x): 2.5 μL • Sample cDNA: 12.5 μL | PCR conditions (cycle # depending on library ): – 98 °C for 30 s – 98 °C for 15 s → 68 °C for 30 s → 72 °C for 40 s (×6 cycles) – 98 °C for 15 s → 72 °C for 60 s (×[qPCR Cq minus 9] cycles) – 72 °C 1 min – 4 °C hold Total cycle # for final PCR: 3 cycles less than the qPCR Ct or Cq of the 1:10 diluted sample |
-
Dispense into 8-well strips, add 12.5 μL CLIP sample + 2.5 μL H 2 O and mix.
-
Perform PCR as indicated above.
3.11.3 SPRI Cleanup Library
-
Add 90 μL AmpureXP beads suspension (do not separate) per 50 μL PCR reaction, mix, incubate at room temperature for 10 min (pipette mix 2–3× during incubation).
-
Magnetically separate, wash beads 2× with 75% EtOH remove the supernatant → air-dry beads 5 min.
-
Resuspend in 20 μL H 2 O, let it sit for 5 min, magnetically separate for 5 min.
-
Transfer 18 μL to new tubes.
3.12 Agarose Gel Electrophoresis and Size-Selection of Library
-
Prepare 3% low-melting temp agarose gel with 1:10,000 SybrSafe in 1× TBE.
3.12.1 Prepare Samples and Run Gel
-
Add 6 μL 6× OrangeG buffer to each sample (18 μL of sample), mix.
-
Load on gel, leave one empty well between samples, 50 bp ladder on both sides of the gel.
-
Run ~95 V for 50 min (longer gives better resolution but larger cut sizes).
3.12.2 Gel-Extract Library from Gel
-
Under blue light illumination, cut gel slice from 175–350 bp and place into 15 mL conical tube.
-
Use fresh razor blades for each sample and keep cross-contamination to minimum.
-
Cut and elute gel using Qiagen MinElute gel extraction kit:
-
Add 6× volumes of Buffer QG to melt gel (e.g., for 100 mg gel, add 600 μL QG).
-
Melt gel at room temp (do not heat) on benchtop (can shake to help melt, but do not vortex).
-
Add 1× volume of original gel of isopropanol and mix well (100 mg gel = 100 μL isopropanol).
-
Load on column (750 μL per spin, can do multiple spins, all spins max speed 1 min).
-
After all sample has been spun through, wash 1× with 500 μL Buffer QG.
-
Add 1× with 750 μL Buffer PE, spin 1 min, pour out flow-through, spin again 2 min max speed.
-
Carefully move column to a new 1.5 mL tube, and air dry for 2 min.
-
Carefully add 12.5 μL Buffer EB directly to the center of the column, incubate for 2 min at room temperature, spin at max speed.
-
3.13 Quantitate Library
-
Quantitate using D1000 DNA Tapes for the Agilent TapeStation.
4 Notes
-
1.
RNase inhibitor addition to lysis buffer (Subheading 3.2.1 ).
Murine RNase inhibitor (NEB) inhibits many endogenous RNase enzymes but does not significantly inhibit RNase I. As such, it can be added either before or after RNase I treatment. For many cell lines (HEK293T, K562) we have observed that this choice does not alter fragmentation or ultimate signal. However, we have observed that for cell types or tissues with moderate to high endogenous RNase activity (e.g., stem cells, differentiated neurons, many tissues), the addition of RNase inhibitor at the initial lysis step is essential to prevent over-fragmentation. We note that the amount of RNase inhibitor may need to be further increased for samples with particularly high RNase activity (e.g., liver or pancreas).
-
2.
Buffer transitions at wash (Subheadings 3.4.1 , 3.4.4 , and others).
We often perform intermediate washes with equal mixtures of the previous and current wash buffer, in order to decrease dramatic changes in buffer composition.
-
3.
Modified adaptor strategy for single-end enhanced CLIP (seCLIP) (Subheadings 3.5.1 , 3.8.1 , and 3.10.1 ).
eCLIP and iCLIP methods utilize the tendency of reverse transcriptase to terminate at the protein-crosslinked RNA nucleotide to enable single-nucleotide resolution of binding sites. In the initial eCLIP adaptor strategy (referred to here as paired-end peCLIP), this position was located at the beginning of the second (paired-end) read in standard Illumina sequencing , which yielded high-quality data but added additional cost. To enable single-end eCLIP (seCLIP), we created a modified adapter strategy that inverted this read structure (Fig. 2a), but used the same highly efficient enzymatic steps (including a 3′ RNA adapter ligation to the RNA fragments, reverse transcription using a melting temperature-optimized primer, and a 3′ ssDNA ligation to the cDNA fragment). Performing both peCLIP and seCLIP on well-characterized splicing regulator RBFOX2 in HEK293T cells, we observed similar read density profiles for individual example binding sites (Fig. 2b) and comparable results in library yield (quantified as the number of PCR cycles necessary to obtain 100 femtomoles of library (eCT)) with biological replicates averaging 12.3 and 12.7 eCT for peCLIP and seCLIP respectively (Fig. 2c). Transcriptome -wide, we observed high correlation of read density within peaks between peCLIP and seCLIP (R 2 = 0.46 and 0.52), equivalent to those observed between biological replicates of peCLIP (R 2 = 0.52) or seCLIP (R 2 = 0.47) (Fig. 2d). Confirming that seCLIP maintains single-nucleotide resolution, we observed the same stereotypical enrichment for the RBFOX family motif (UGCAUG) enriched at the −6 and −2 positions relative to read start positions (Fig. 2e). Thus, this modified seCLIP adapter strategy enables the same high-quality eCLIP data generation amenable to sequencing with standard single-end, 50 bp chemistry, decreasing cost. Further, the use of standard Illumina barcodes in seCLIP enables pooling with other standard Illumina RNA-seq or other high-throughput sequencing libraries.
Fig. 2 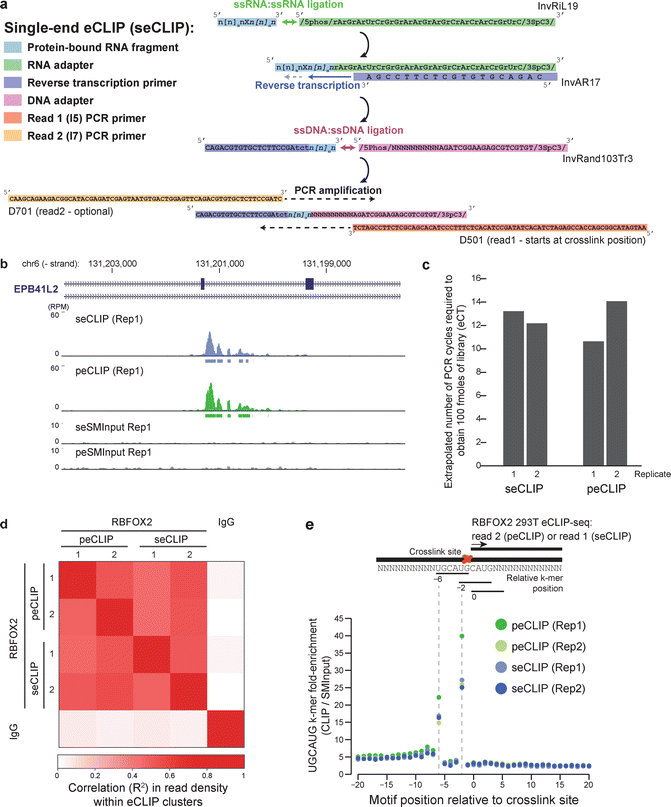
Modified adaptor strategy for single-end enhanced CLIP (seCLIP). (a) Schematic of adaptor sequences used in seCLIP . (b) Read density (shown as reads per million; RPM) observed in original, paired-end eCLIP (peCLIP) and seCLIP for RBFOX2 in HEK293T cells at EPB41L2 exon 13-14. Boxes below tracks indicate significantly enriched peaks after input normalization. (c) Required amplification observed using seCLIP and peCLIP adaptor strategies for two biological replicates profiling RBFOX2 in HEK293T. (d) Heatmap indicates correlation across experiments for read fold-enrichment in CLIP versus input, considering peaks identified in indicated experiments (y-axis). (e) Plot indicates enrichment for RBFOX2 binding motif UGCAUG at indicated positions around read start positions
-
4.
Decreasing contamination introduced by nitrocellulose membrane sources (Subheading 3.6.4 ).
In 102 K562 and HepG2 eCLIP experiments, an average of 84% of reads uniquely or multiply mapped to the human genome, respectively [11]. However, in preliminary experiments in cell types with decreased RNA yield after membrane transfer (motor neurons (MN) and neural progenitor cells (NPCs) derived from human embryonic stem cells), we observed in some cases more than 90% of sequenced reads were not mapped to the human genome. Using SOAP-denovo [13] to de novo assemble the unmapped reads, we assembled multiple contigs that were queried against the NR database and showed >99% identity to Acinetobacter johnsonii XBB1 (CP010350.1). Re-mapping these eCLIP datasets revealed millions of reads in many datasets mapping throughout CP010350.1, confirming this specific species as a major contamination source (Fig. 3a). We noted that reads mapping to CP010350.1 had proper CLIP adapter structure, indicating that contamination was likely occurring prior to the 3′ linker ligation .
Fig. 3 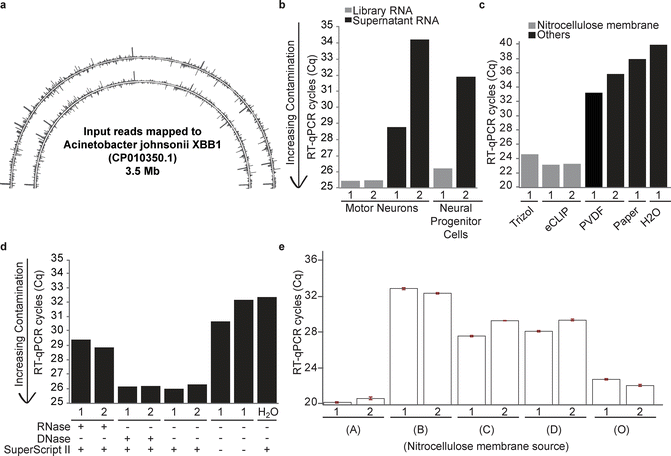
Genome of Acinetobacter johnsonii XBB1 in nitrocellulose membranes detected by RT—qPCR. (a) Sequencing reads from two eCLIP input libraries mapped to Acinetobacter johnsonii XBB1 (CP010350.1). (b) Bars indicate Cq from RT—qPCR performed using CP010350.1-specific qPCR primers on eCLIP RNA (Subheading 3.7.3) and supernatant samples (Subheading 3.4.1) from indicated cell types. Lower Cq reflects higher CP010350.1 signal. Numbers indicate replicate experiments. (c) Bars indicate RT—qPCR Cq for CP010350.1 from RNA isolated from nitrocellulose membranes via indicated method, PVDF membranes (RNA isolation by Trizol), paper (RNA isolation by Trizol), and H2O (Trizol extraction). (d) Bars indicate RT—qPCR Ct for CP010350.1 from RNA extracted from size-matched nitrocellulose membranes, in technical replicates. Symbols below indicate samples that were either RNase or DNase treated, and those with and without RT enzyme added. (e) Bars indicate RT—qPCR Ct for CP010350.1 from RNA extracted from nitrocellulose membrane samples from five sources in technical replicates as follows: (A) commercial source A, (B) ThermoFisher iBlot (IB23001 lot 2NR26016-01), (C) GE Amersham Protran Premium (13600117 lot G6552142), (D) GE Amersham Protran Premium (1060008 lot G9931040), and (O) original commercial source. Error bars indicate standard deviation from RT—qPCR triplicate measurements
In order to modify eCLIP to ameliorate this issue, we set out to identify the source of this contamination by performing RT-qPCR using primers designed against regions of CP010350.1 with high read density. To first confirm that this contamination was not present in initial samples, we extracted RNA (Trizol LS) from supernatant remaining after immunoprecipitation (Subheading 3.4.1) in addition to standard post-membrane transfer and isolation (Subheading 3.7.3) and performed RT-qPCR for bacterial RNA. We observed more than tenfold increased bacterial RNA signal in the membrane-isolated RNA as compared to supernatant RNA from the equivalent number of cells, indicating that the contamination was not present during tissue culture and was introduced during the IP/Western stage (Fig. 3b). Similar RT-qPCR assays performed after RNA isolation on various buffers or enzyme mixes used failed to identify significant contamination (data not shown).
The observation that these reads were only present in input but not CLIP samples (despite input often having more than 100-fold more RNA recovery and library yield [12]), implicated the difference in RNA adaptor ligation of CLIP (3′ RNA adaptor ligation on-bead, before the protein electrophoresis step) versus input (3′ RNA adaptor ligation after RNA isolation off of membranes) RNA. Surprisingly, we found that RNA extraction (Trizol) of nitrocellulose membrane alone yielded RT-qPCR signal similar to our contaminated libraries, in contrast to the lower signal observed after RNA isolation from PVDF membranes, Whatman and other lab paper, or negative controls (Fig. 3c). We observed identical results with freshly ordered membrane stock (data not shown). To further explore the nature of the contamination, we synthesized cDNA from either RNase or DNase-treated membrane samples and repeated the RT-qPCR assay. This indicated that the contamination was likely RNA, as the RT-qPCR signal was sensitive to both RNase and the no-RT control, but not DNase (Fig. 3d). The strand-specific signal observed in reads similarly implicated RNA contamination (Fig. 3a).
As the nitrocellulose transfer provides key specificity for isolating RNA crosslinked to protein, we set out to identify optimized alternative sources that had decreased RNA background. We obtained four additional nitrocellulose membrane sources (A), (B) ThermoFisher iBlot2 (IB23001 lot 2NR26016-01), (C) GE Amersham Protran Premium (13600117 lot G6552142), and (D) GE Amersham Protran Premium (1060008 lot G9931040), in addition to our original commercial source (O). For each, we performed RNA isolation followed by the bacterial RNA RT-qPCR, and observed that whereas O and A showed similar CP010350.1 contamination, B, C, and D did not (Fig. 3e). When we prepared libraries according to our standard protocol for eCLIP input samples, we observed that library yields reflected these results, with B, C, and D showing the least amount of overall contamination (data not shown). Importantly, we observed no difference in RNA or library yield when we prepared standard eCLIP input libraries for two protein size ranges of roughly equal membrane size (10–50 kDa and 50–225 kDa) for multiple membrane types. Thus, these results indicate that testing of nitrocellulose membranes enables optimization of eCLIP by removing substantial background contamination. Our results identify (B) ThermoFisher iBlot and (C) GE Amersham Protran Premium (1060008) membranes as options which show a dramatic decrease in contamination for sensitive eCLIP experiments without altering true library yield. Although yielding equally low contamination, (D) GE Amersham Protran Premium (13600117) is “trial-size” packaging and generally commercially unavailable for large-scale use. Other sources can be tested using the RT-qPCR method described here to determine whether they are of sufficiently low background for use in eCLIP.


References
Morris KV, Mattick JS (2014) The rise of regulatory RNA. Nat Rev Genet 15(6):423–437. doi:10.1038/nrg3722
Quinn JJ, Chang HY (2016) Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17(1):47–62. doi:10.1038/nrg.2015.10
Jonas S, Izaurralde E (2015) Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet 16(7):421–433. doi:10.1038/nrg3965
Glisovic T, Bachorik JL, Yong J, Dreyfuss G (2008) RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett 582(14):1977–1986. doi:10.1016/j.febslet.2008.03.004
Gerstberger S, Hafner M, Tuschl T (2014) A census of human RNA-binding proteins. Nat Rev Genet 15(12):829–845. doi:10.1038/nrg3813
Licatalosi DD, Darnell RB (2010) RNA processing and its regulation: global insights into biological networks. Nat Rev Genet 11(1):75–87. doi:10.1038/nrg2673
Konig J, Zarnack K, Luscombe NM, Ule J (2011) Protein-RNA interactions: new genomic technologies and perspectives. Nat Rev Genet 13(2):77–83. doi:10.1038/nrg3141
Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB (2003) CLIP identifies Nova-regulated RNA networks in the brain. Science 302(5648):1212–1215. doi:10.1126/science.1090095
Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T (2010) Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141(1):129–141. doi:10.1016/j.cell.2010.03.009
Zhang C, Darnell RB (2011) Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol 29(7):607–614. doi:10.1038/nbt.1873
Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J (2010) iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17(7):909–915. doi:10.1038/nsmb.1838
Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, Stanton R, Rigo F, Guttman M, Yeo GW (2016) Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13(6):508–514. doi:10.1038/nmeth.3810
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J, Wu G, Zhang H, Shi Y, Liu Y, Yu C, Wang B, Lu Y, Han C, Cheung DW, Yiu SM, Peng S, Xiaoqian Z, Liu G, Liao X, Li Y, Yang H, Wang J, Lam TW, Wang J (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1(1):18. doi:10.1186/2047-217X-1-18
Acknowledgments
The authors would like to thank members of the Yeo lab for insightful discussions and critical reading of the manuscript, particularly S. Aigner. This work was supported by grants from the National Institute of Health [HG004659, HG007005, and NS075449 to G.W.Y.]. E.L.V.N. is a Merck Fellow of the Damon Runyon Cancer Research Foundation [DRG-2172-13]. G.A.P. is supported by the National Science Foundation Graduate Research Fellowship. G.W.Y. is an Alfred P. Sloan Research Fellow.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made. The images or other third party material in this chapter are included in the chapter’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2017 The Author(s)
About this protocol
Cite this protocol
Van Nostrand, E.L. et al. (2017). Robust, Cost-Effective Profiling of RNA Binding Protein Targets with Single-end Enhanced Crosslinking and Immunoprecipitation (seCLIP). In: Shi, Y. (eds) mRNA Processing. Methods in Molecular Biology, vol 1648. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7204-3_14
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7204-3_14
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-7203-6
Online ISBN: 978-1-4939-7204-3
eBook Packages: Springer Protocols