
Metagenomic Analysis of Virioplankton of the Subtropical Jiulong River Estuary, China


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Sequencing
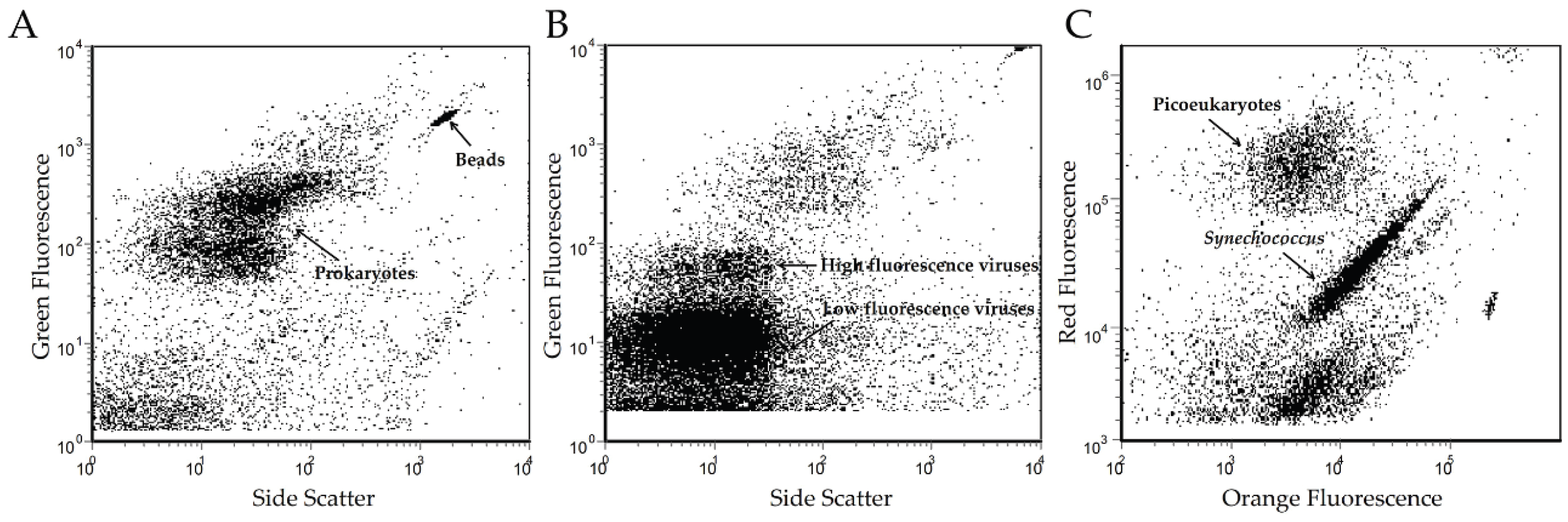
2.2. Microbial Enumeration
2.3. Bioinformatics Analysis
3. Results
3.1. Environmental Characteristics

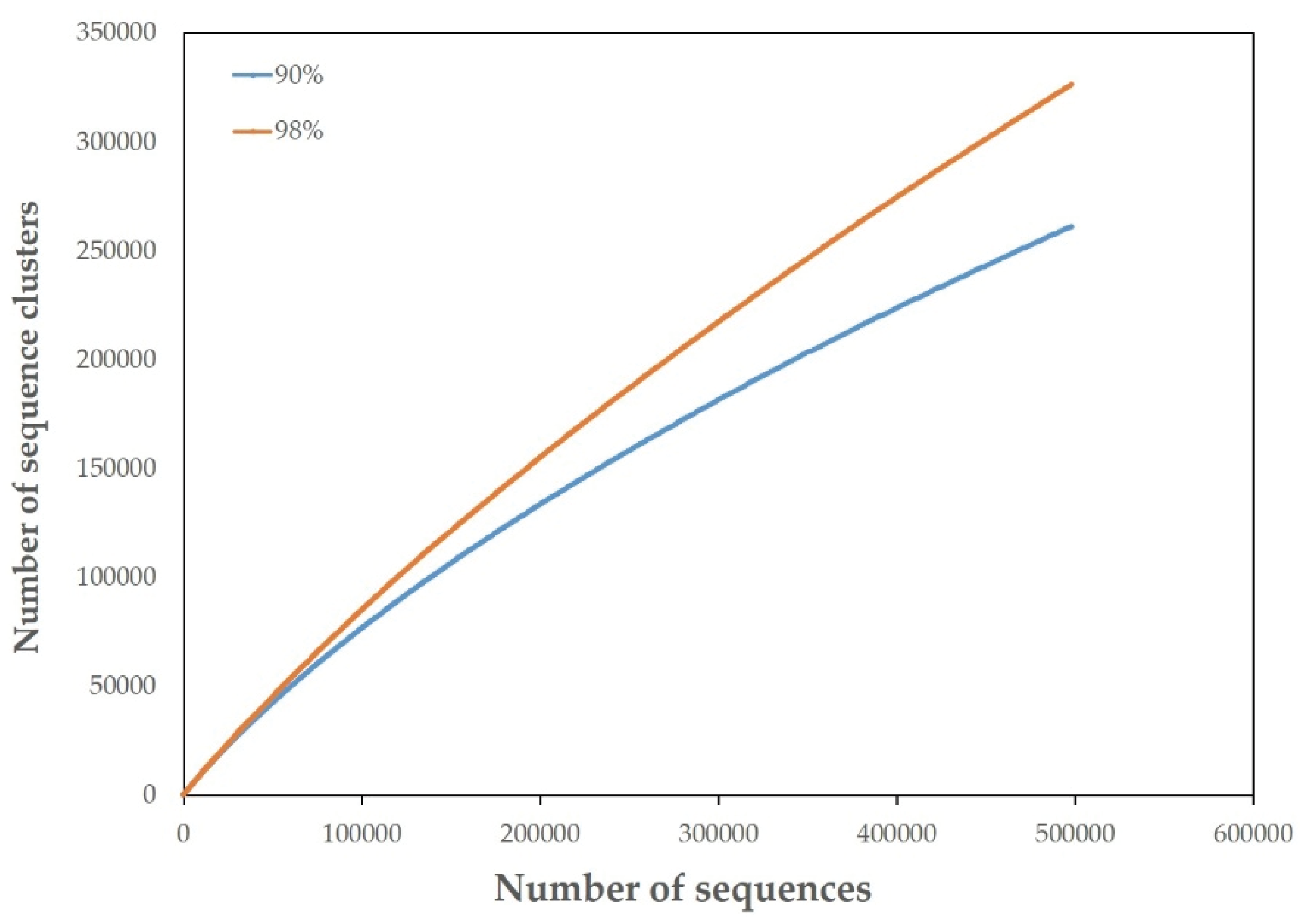
3.2. Overview of the JRE Virome

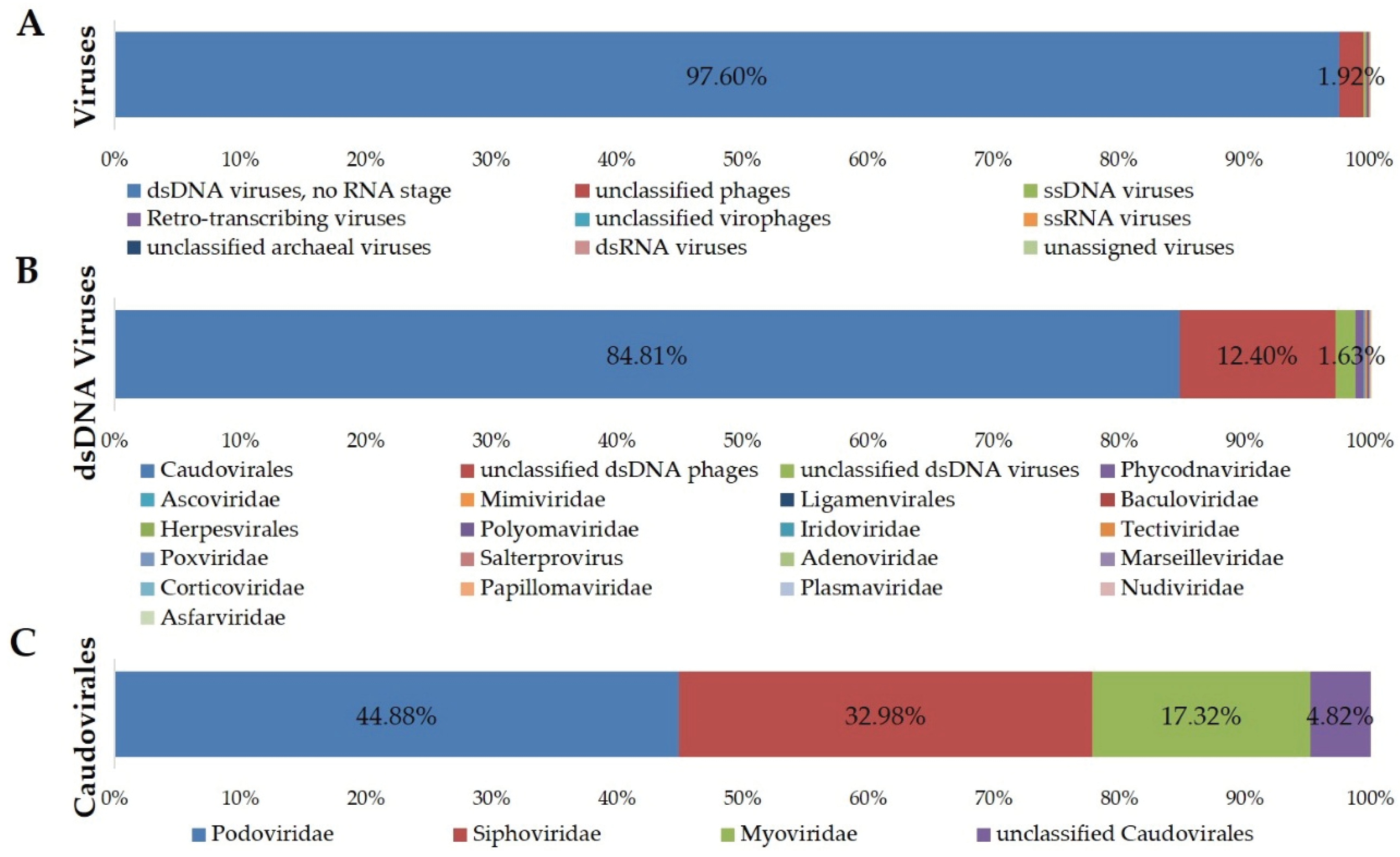
3.3. Taxonomic Composition

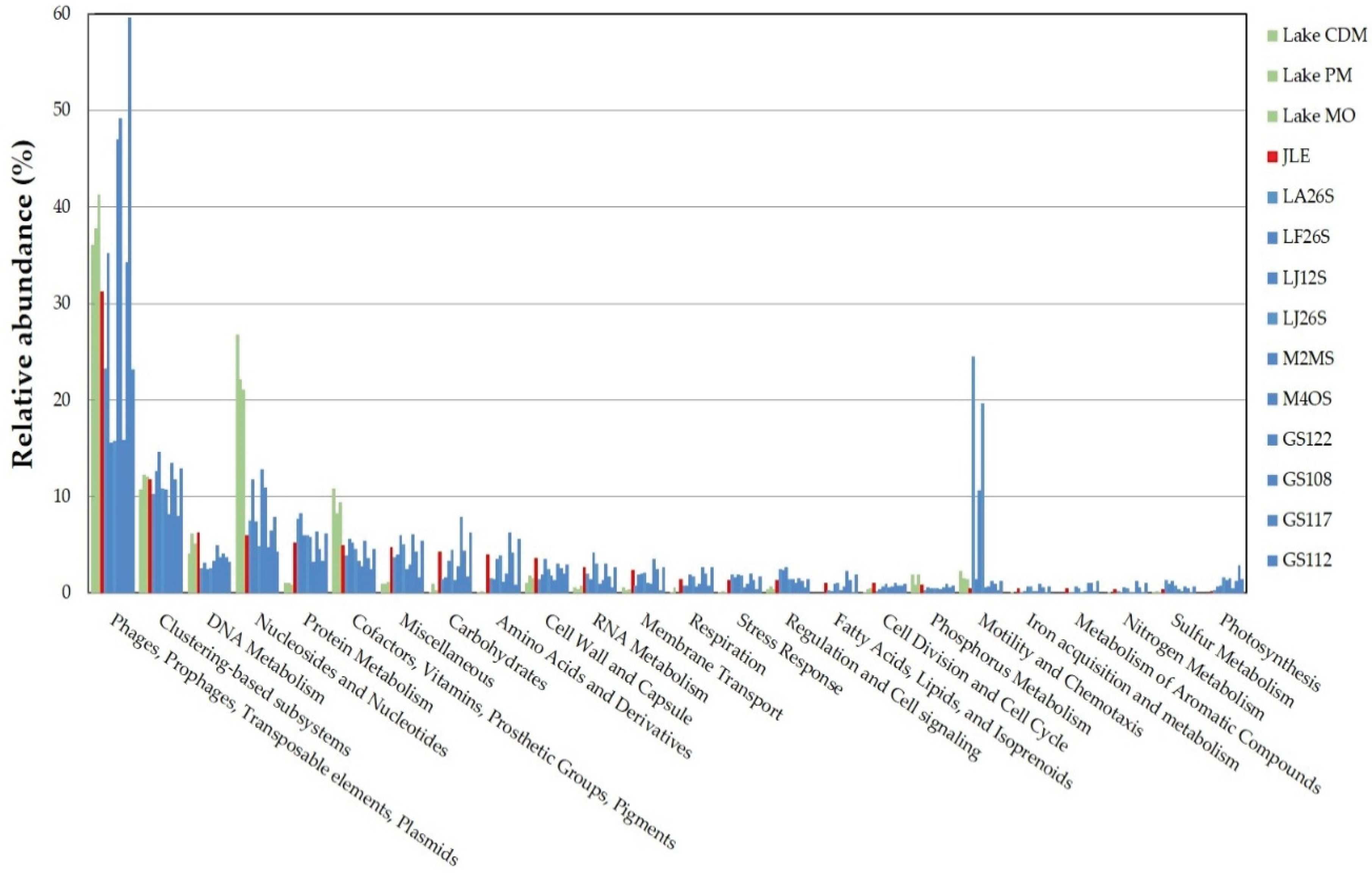
3.4. Functional Analysis

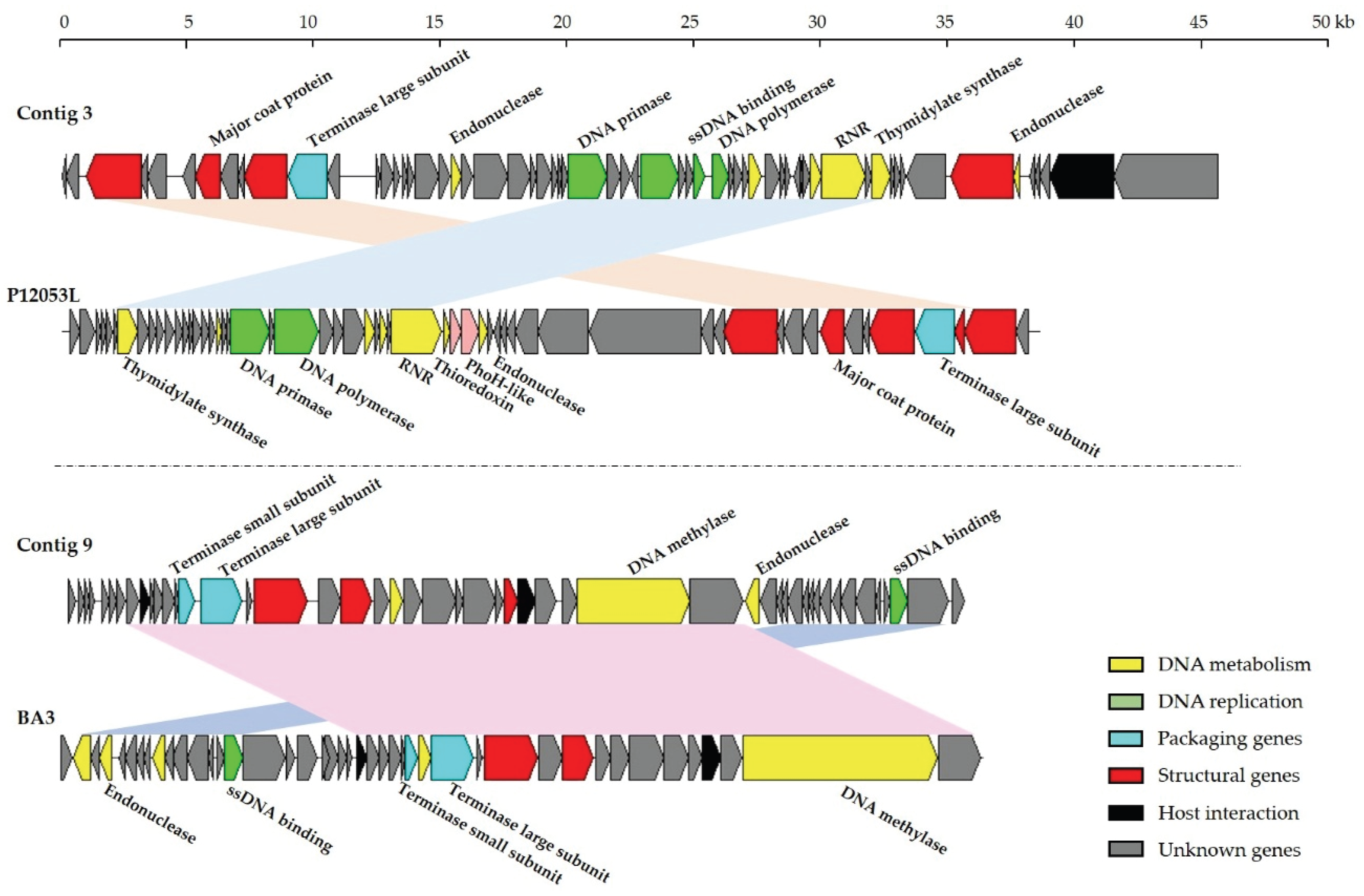
3.5. Contig Analysis

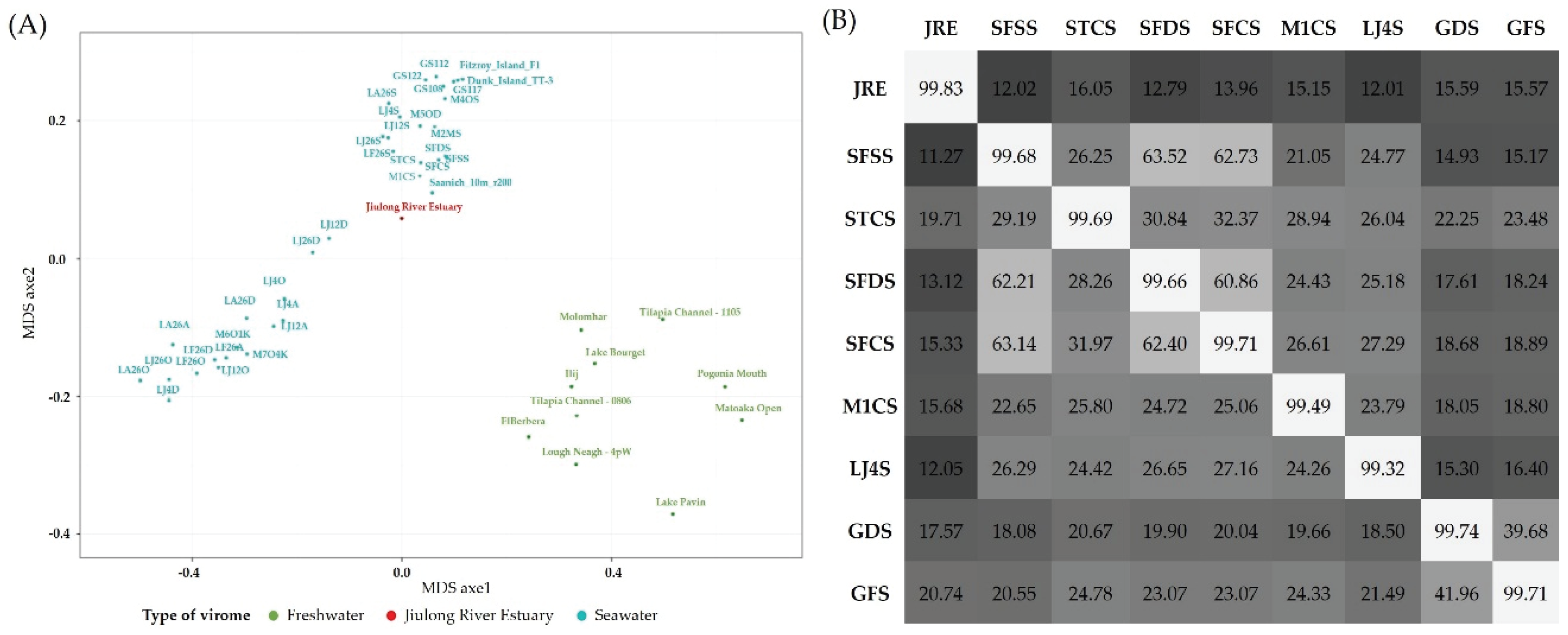
3.6. Comparison with Other Viromes

4. Discussion
4.1. The JRE Virome
4.2. Impacts of Marine Water on the Composition of the JRE Virome
4.3. Impacts of River Outflow on the Composition of the JRE Virome
4.4. Methodological Considerations and Limitations
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, S.; Miki, T.; Noble, R.; Peduzzi, P.; Wilhelm, S. Viruses in aquatic ecosystems: Important advancements of the last 20 years and prospects for the future in the field of microbial oceanography and limnology. Adv. Oceanogr. Limnol. 2010, 1, 97–141. [Google Scholar] [CrossRef]
- Thingstad, T.F. Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol. Oceanogr. 2000, 45, 1320–1328. [Google Scholar] [CrossRef]
- Rohwer, F.; Thurber, R.V. Viruses manipulate the marine environment. Nature 2009, 459, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M. Marine viruses: Truth or dare. Ann. Rev. Mar. Sci. 2012, 4, 425–448. [Google Scholar] [CrossRef]
- Brum, J.R.; Sullivan, M.B. Rising to the challenge: Accelerated pace of discovery transforms marine virology. Nat. Rev. Microbiol. 2015. [Google Scholar] [CrossRef]
- Lindell, D.; Jaffe, J.D.; Johnson, Z.I.; Church, G.M.; Chisholm, S.W. Photosynthesis genes in marine viruses yield proteins during host infection. Nature 2005, 438, 86–89. [Google Scholar] [PubMed]
- Thompson, L.R.; Zeng, Q.; Kelly, L.; Huang, K.H.; Singer, A.U.; Stubbe, J.; Chisholm, S.W. Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc. Natl. Acad. Sci. USA 2011, 108, E757–E764. [Google Scholar] [CrossRef] [PubMed]
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wei, W.; Cai, L. The fate and biogeochemical cycling of viral elements. Nat. Rev. Microbiol. 2014, 12, 850–851. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Herndl, G.J.; Hansell, D.A.; Benner, R.; Kattner, G.; Wilhelm, S.W.; Kirchman, D.L.; Weinbauer, M.G.; Luo, T.; Chen, F.; et al. Microbial production of recalcitrant dissolved organic matter: Long-term carbon storage in the global ocean. Nat. Rev. Microbiol. 2010, 8, 593–599. [Google Scholar] [CrossRef]
- Breitbart, M.; Salamon, P.; Andresen, B.; Mahaffy, J.M.; Segall, A.M.; Mead, D.; Azam, F.; Rohwer, F. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. USA 2002, 99, 14250–14255. [Google Scholar] [CrossRef] [PubMed]
- Angly, F.E.; Felts, B.; Breitbart, M.; Salamon, P.; Edwards, R.A.; Carlson, C.; Chan, A.M.; Haynes, M.; Kelley, S.; Liu, H.; et al. The marine viromes of four oceanic regions. PLoS Biol. 2006, 4, e368. [Google Scholar] [CrossRef] [PubMed]
- Green, J.C.; Rahman, F.; Saxton, M.A.; Williamson, K.E. Metagenomic assessment of viral diversity in lake matoaka, a temperate, eutrophic freshwater lake in southeastern Virginia, USA. Aquat. Microbial. Ecol. 2015, 75, 117–128. [Google Scholar] [CrossRef]
- Tseng, C.H.; Chiang, P.W.; Shiah, F.K.; Chen, Y.L.; Liou, J.R.; Hsu, T.C.; Maheswararajah, S.; Saeed, I.; Halgamuge, S.; Tang, S.L. Microbial and viral metagenomes of a subtropical freshwater reservoir subject to climatic disturbances. ISME J. 2013, 7, 2374–2386. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Robin, A.; Ravet, V.; Personnic, S.; Theil, S.; Colombet, J.; Sime-Ngando, T.; Debroas, D. Assessing the diversity and specificity of two freshwater viral communities through metagenomics. PLoS ONE 2012, 7, e33641. [Google Scholar]
- Aguirre de Carcer, D.; Lopez-Bueno, A.; Pearce, D.A.; Alcami, A. Biodiversity and distribution of polar freshwater DNA viruses. Sci. Adv. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, B.; Freeborough, M.J.; Maree, H.J.; Celton, J.M.; Rees, D.J.; Burger, J.T. Deep sequencing analysis of viruses infecting grapevines: Virome of a vineyard. Virology 2010, 400, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 2003, 185, 6220–6223. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.S.; Kapoor, A.; Lukashov, V.V.; Simmonds, P.; Hecht, F.; Delwart, E. New DNA viruses identified in patients with acute viral infection syndrome. J. Virol. 2005, 79, 8230–8236. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.; Suedmeyer, W.K.; Wheeler, E.; Gulland, F.; Breitbart, M. Novel anellovirus discovered from a mortality event of captive California sea lions. J. Gen. Virol 2009, 90, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, T.; Patterson, M.; Richardson, P.M.; Wommack, K.E.; Young, M.; Mead, D. Assembly of viral metagenomes from Yellowstone hot springs. Appl. Environ. Microbiol. 2008, 74, 4164–4174. [Google Scholar] [CrossRef]
- Lopez-Bueno, A.; Tamames, J.; Velazquez, D.; Moya, A.; Quesada, A.; Alcami, A. High diversity of the viral community from an Antarctic lake. Science 2009, 326, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Aw, T.G.; Teal, T.K.; Rose, J.B. Metagenomic investigation of viral communities in ballast water. Environ. Sci. Technol. 2015, 49, 8396–8407. [Google Scholar] [CrossRef] [PubMed]
- Martinez Martinez, J.; Swan, B.K.; Wilson, W.H. Marine viruses, a genetic reservoir revealed by targeted viromics. ISME J. 2014, 8, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Wommack, K.E.; Bhavsar, J.; Ravel, J. Metagenomics: Read length matters. Appl. Environ. Microbiol. 2008, 74, 1453–1463. [Google Scholar] [CrossRef]
- Brum, J.R.; Ignacio-Espinoza, J.C.; Roux, S.; Doulcier, G.; Acinas, S.G.; Alberti, A.; Chaffron, S.; Cruaud, C.; de Vargas, C.; Gasol, J.M.; et al. Patterns and ecological drivers of ocean viral communities. Science 2015, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helton, R.R.; Wommack, K.E. Seasonal dynamics and metagenomic characterization of estuarine viriobenthos assemblages by randomly amplified polymorphic DNA PCR. Appl. Environ. Microbiol. 2009, 75, 2259–2265. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, C.S.; Herfort, L.; Zuber, P.; Baptista, A.M.; Crump, B.C. Spatial variability overwhelms seasonal patterns in bacterioplankton communities across a river to ocean gradient. ISME J. 2012, 6, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.L.; Sullivan, M.B. The Pacific Ocean Virome (POV): A marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS ONE 2013, 8, e57355. [Google Scholar] [CrossRef] [PubMed]
- Shan, D.; Wei, G.; Li, M.; Wang, W.; Li, X.; Gao, Z.; Shao, Z. Distribution and diversity of bacterioplankton communities in subtropical seawater around Xiamen island, China. Microbiol. Res. 2015, 175, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Marie, D.; Brussaard, C.P.D.; Thyrhaug, R.; Bratbak, G.; Vaulot, D. Enumeration of marine viruses in culture and natural samples by flow cytometry. Appl. Environ. Microbiol. 1999, 65, 45–52. [Google Scholar] [PubMed]
- Brussaard, C.P. Optimization of procedures for counting viruses by flow cytometry. Appl. Environ. Microbiol. 2004, 70, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.Z.; Yang, Y.H.; Koshikawa, H.; Watanabe, M. Influence of hydrographie conditions on picoplankton distribution in the East China Sea. Aquat. Microbial. Ecol. 2002, 30, 37–48. [Google Scholar] [CrossRef]
- EXPO32; v1.2; Beckman Coulter: Miami, FL, USA, 2001.
- Niu, B.; Fu, L.; Sun, S.; Li, W. Artificial and natural duplicates in pyrosequencing reads of metagenomic data. BMC Bioinform. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Mothur. Available online: http://www.mothur.org/ (accessed on 30 January 2016).
- Roux, S.; Faubladier, M.; Mahul, A.; Paulhe, N.; Bernard, A.; Debroas, D.; Enault, F. Metavir: A web server dedicated to virome analysis. Bioinformatics 2011, 27, 3074–3075. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. Genemarks: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; van Zyl, L.; de Maayer, P.; Rubagotti, E.; Rybicki, E.; Tuffin, M.; Cowan, D.A. Metagenomic analysis of the viral community in Namib Desert hypoliths. Environ. Microbiol. 2015, 17, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Steward, G.F.; Montiel, J.L.; Azam, F. Genome size distributions indicate variability and similarities among marine viral assemblages from diverse environments. Limnol. Oceanogr. 2000, 45, 1697–1706. [Google Scholar] [CrossRef]
- Angly, F.E.; Willner, D.; Prieto-Davo, A.; Edwards, R.A.; Schmieder, R.; Vega-Thurber, R.; Antonopoulos, D.A.; Barott, K.; Cottrell, M.T.; Desnues, C.; et al. The GAAS metagenomic tool and its estimations of viral and microbial average genome size in four major biomes. PLoS Comput. Biol. 2009, 5, e1000593. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.J.; Allen, L.Z.; Lorenzi, H.A.; Fadrosh, D.W.; Brami, D.; Thiagarajan, M.; McCrow, J.P.; Tovchigrechko, A.; Yooseph, S.; Venter, J.C. Metagenomic exploration of viruses throughout the Indian Ocean. PLoS ONE 2012, 7, e42047. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Jang, H.; Oh, H.M.; Cho, J.C. Complete genome sequence of Celeribacter bacteriophage P12053L. J. Virol. 2012, 86, 8339–8340. [Google Scholar] [CrossRef]
- Winter, C.; Garcia, J.A.; Weinbauer, M.G.; DuBow, M.S.; Herndl, G.J. Comparison of deep-water viromes from the Atlantic Ocean and the Mediterranean Sea. PLoS ONE 2014, 9, e100600. [Google Scholar]
- Chow, C.E.; Winget, D.M.; White, R.A., 3rd; Hallam, S.J.; Suttle, C.A. Combining genomic sequencing methods to explore viral diversity and reveal potential virus-host interactions. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.M.; Rappe, M.S.; Connon, S.A.; Vergin, K.L.; Siebold, W.A.; Carlson, C.A.; Giovannoni, S.J. SAR11 clade dominates ocean surface bacterioplankton communities. Nature 2002, 420, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Temperton, B.; Thrash, J.C.; Schwalbach, M.S.; Vergin, K.L.; Landry, Z.C.; Ellisman, M.; Deerinck, T.; Sullivan, M.B.; Giovannoni, S.J. Abundant SAR11 viruses in the ocean. Nature 2013, 494, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Oh, H.M.; Kang, D.; Cho, J.C. Genome of a SAR116 bacteriophage shows the prevalence of this phage type in the oceans. Proc. Natl. Acad. Sci. USA 2013, 110, 12343–12348. [Google Scholar] [CrossRef] [PubMed]
- Efrony, R.; Loya, Y.; Bacharach, E.; Rosenberg, E. Phage therapy of coral disease. Coral Reefs 2007, 26, 7–13. [Google Scholar] [CrossRef]
- Velammal, A.; Aiyamperumal, B.; Venugopalan, V.K.; Ajmalkhan, S. Distribution of Pseudomonas aeruginosa in pondicherry coastal environs. Indian J. Mar. Sci. 1994, 23, 239–241. [Google Scholar]
- Mates, A. The significance of testing for Pseudomonas aeruginosa in recreational seawater beaches. Microbios 1992, 71, 89–93. [Google Scholar] [PubMed]
- Kimata, N.; Nishino, T.; Suzuki, S.; Kogure, K. Pseudomonas aeruginosa isolated from marine environments in Tokyo Bay. Microb. Ecol. 2004, 47, 41–47. [Google Scholar] [CrossRef]
- Jiao, N.Z.; Zhao, Y.L.; Luo, T.W.; Wang, X.L. Natural and anthropogenic forcing on the dynamics of virloplankton in the Yangtze river estuary. J. Mar. Biol. Assoc. UK 2006, 86, 543–550. [Google Scholar] [CrossRef]
- Liu, L.; Yang, J.; Zhang, Y. Genetic diversity patterns of microbial communities in a subtropical riverine ecosystem (Jiulong River, southeast China). Hydrobiologia 2011, 678, 113–125. [Google Scholar] [CrossRef]
- Cai, L.; Yang, Y.; Jiao, N.; Zhang, R. Evaluation of tangential flow filtration for the concentration and separation of bacteria and viruses in contrasting marine environments. PLoS ONE 2015, 10, e0136741. [Google Scholar] [CrossRef]
- John, S.G.; Mendez, C.B.; Deng, L.; Poulos, B.; Kauffman, A.K.; Kern, S.; Brum, J.; Polz, M.F.; Boyle, E.A.; Sullivan, M.B. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ. Microbiol. Rep. 2011, 3, 195–202. [Google Scholar] [CrossRef]
- Brum, J.R.; Schenck, R.O.; Sullivan, M.B. Global morphological analysis of marine viruses shows minimal regional variation and dominance of non-tailed viruses. ISME J. 2013, 7, 1738–1751. [Google Scholar] [CrossRef] [PubMed]
- Bellas, C.M.; Anesio, A.M.; Barker, G. Analysis of virus genomes from glacial environments reveals novel virus groups with unusual host interactions. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, L.; Zhang, R.; He, Y.; Feng, X.; Jiao, N. Metagenomic Analysis of Virioplankton of the Subtropical Jiulong River Estuary, China. Viruses 2016, 8, 35. https://doi.org/10.3390/v8020035
Cai L, Zhang R, He Y, Feng X, Jiao N. Metagenomic Analysis of Virioplankton of the Subtropical Jiulong River Estuary, China. Viruses. 2016; 8(2):35. https://doi.org/10.3390/v8020035
Chicago/Turabian StyleCai, Lanlan, Rui Zhang, Ying He, Xiaoyuan Feng, and Nianzhi Jiao. 2016. "Metagenomic Analysis of Virioplankton of the Subtropical Jiulong River Estuary, China" Viruses 8, no. 2: 35. https://doi.org/10.3390/v8020035